

GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide
For Research Use Only. Not for use in diagnostic procedures.
Information in this document is subject to change without notice.
APPLIED BIOSYSTEMS DISCLAIMS ALL WARRANTIES WITH RESPECT TO THIS DOCUMENT, EXPRESSED OR
IMPLIED, INCLUDING BUT NOT LIMITED TO THOSE OF MERCHANTABILITY OR FITNESS FOR A PARTICULAR
PURPOSE. TO THE FULLEST EXTENT ALLOWED BY LAW, IN NO EVENT SHALL APPLIED BIOSYSTEMS BE
LIABLE, WHETHER IN CONTRACT, TORT, WARRANTY, OR UNDER ANY STATUTE OR ON ANY OTHER BASIS FOR
SPECIAL, INCIDENTAL, INDIRECT, PUNITIVE, MULTIPLE OR CONSEQUENTIAL DAMAGES IN CONNECTION WITH
OR ARISING FROM THIS DOCUMENT, INCLUDING BUT NOT LIMITED TO THE USE THEREOF, WHETHER OR NOT
FORESEEABLE AND WHETHER OR NOT APPLIED BIOSYSTEMS IS ADVISED OF THE POSSIBILITY OF SUCH
DAMAGES.
NOTICE TO PURCHASER: DISCLAIMER OF LICENSE
Purchase of this software product alone does not imply any license under any process, instrument or other apparatus,
system, composition, reagent or kit rights under patent claims owned or otherwise controlled by Applied Biosystems, either
expressly, or by estoppel.
GeneMapper Software has not undergone specific developmental validation for human identification applications. Human
identification laboratories analyzing single-source or parentage samples which choose to use GeneMapper Software for data
analysis should perform their own developmental validation studies.
The AFLP process is covered by patents owned by Keygene N.V.
TRADEMARKS:
Applied Biosystems, AB (Design), ABI PRISM, GeneMapper, GeneScan, Primer Focus, and SNaPshot are registered trademarks,
and GeneScan and SNPlex are trademarks of Applied Biosystems or its affiliates in the U.S. and/or certain other countries.
AFLP is a registered trademark of Keygene N.V.
This product includes software developed by the Apache Software Foundation (http://www.apache.org/). Copyright © 1999-2000
The Apache Software Foundation. All rights reserved.
This product includes software developed by the ExoLab Project (http://www. exolab.org/). Copyright 2000 © Intalio Inc. All
rights reserved.
JNIRegistry is Copyright © 1997 Timothy Gerard Endres, ICE Engineering, Inc., http://www.trustice.com.
Oracle is a registered trademark of Oracle Corporation.
All other trademarks are the sole property of their respective owners.
© Copyright 2009, Applied Biosystems. All rights reserved.
Part Number 4403673 Rev. A
04/2009

Contents
GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide iii
Preface v
How to Use This Guide . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . v
How to Obtain More Information . . . . . . . . . . . . . . . . . . . . . . . . . . vi
How to Obtain Support . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . viii
Chapter 1 Process Quality Values and Basic
Troubleshooting 1
Diagnosing and Resolving Basic Problems . . . . . . . . . . . . . . . . . . . 2
Common Troubleshooting Procedures . . . . . . . . . . . . . . . . . . . 5
ADO (Allele Display Overflow) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 8
AE (Allele Edit) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 8
AN (Allele Number) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9
BD (Broad Peak) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9
BIN (Out of Bin Allele) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10
CC (Control Concordance) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11
DP (Double Peak) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12
GQ (Genotype Quality) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13
LPH (Low Peak Height) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15
MNF (Matrix Not Found) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
NB (Narrow Bin) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18
OBA (One Basepair Allele) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18
OS (Offscale) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 19
OVL (Overlap) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20
PHR (Peak Height Ratio) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21
SFNF (Sample File Not Found) . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22
SHP (Sharp Peak) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23
SNF (Size Standard Not Found) . . . . . . . . . . . . . . . . . . . . . . . . . . . 23
Contents
iv GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide
SP (Split Peak) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24
SPA (Single Peak Artifact) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24
SPU (Spectral Pull-Up) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25
SQ (Sizing Quality) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25
XTLK (Cross Talk) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 32
Chapter 2 SNPlex
™
System Troubleshooting 33
Overview . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 34
Chapter 3 Algorithms 35
Genotyping Algorithms . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 36
Peak Detection . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 38
Optimizing Peak Detection Sensitivity . . . . . . . . . . . . . . . . . . . . . 41
Example 1: Reducing Window Size . . . . . . . . . . . . . . . . . . . . 41
Example 2: Reducing Window Size/Increasing Polynomial Degree
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 42
Example 3: Extreme Settings . . . . . . . . . . . . . . . . . . . . . . . . . 43
Slope Thresholds for Peak Start/End Parameters . . . . . . . . . . . . 44
Slope Threshold Example . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 45
Size-Matching/Size-Calling Algorithm . . . . . . . . . . . . . . . . . . . . . . 46
Size-Calling Methods (Classic and Advanced Modes) . . . . . . . . . 47
Least Squares Method . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 47
Cubic Spline Interpolation Method . . . . . . . . . . . . . . . . . . . . . 49
Local Southern Method . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 50
Global Southern Method . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 52
Allele-Calling Algorithms . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 53
Microsatellite Analysis Methods . . . . . . . . . . . . . . . . . . . . . . . 54
SNPlex
™
System Analysis Methods . . . . . . . . . . . . . . . . . . . . 55
Glossary 59
Index 67

GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide v
Preface
How to Use This Guide
Purpose of This
Guide
This guide describes the function of the Process Quality Values
(PQV) for the supported analyses of the GeneMapper
®
Software,
explains the fundamental algorithms used by the software, and
provides basic troubleshooting techniques.
Audience This guide is intended for trained laboratory personnel. Applied
Biosystems is not liable for damage or injury that results from use of
this guide by unauthorized or untrained parties.
Assumptions This guide assumes that:
• You have installed GeneMapper
®
Software Version 4.1 as described
in the
GeneMapper
®
Software Version 4.1 Installation and
Administration Guide
(PN 4403614).
• You have a working knowledge of the Microsoft
®
Windows
®
operating system.
Te xt Co nv e nt io n s This guide uses the following conventions:
• Bold indicates user action. For example:
Type 0, then press Enter for each of the remaining fields.
• Italic text indicates new or important words and is also used for
emphasis. For example:
Before analyzing, always prepare fresh matrix.
• A right arrow bracket () separates successive commands you
select from a drop-down or shortcut menu. For example:
Select FileOpenSpot Set.
Right-click the sample row, then select View FilterView All.

Preface
How to Obtain More Information
vi GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide
User Attention
Words
Two user attention words appear in Applied Biosystems user
documentation. Each word implies a particular level of observation
or action as described below:
Note: Provides information that may be of interest or help but is not
critical to the use of the product.
IMPORTANT! Provides information that is necessary for proper
instrument operation, accurate chemistry kit use, or safe use of a
chemical.
Examples of the user attention words appear below:
Note: The size of the column affects the run time.
Note: The Calibrate function is also available in the Control
Console.
IMPORTANT! To verify your client connection to the database, you
need a valid Oracle user ID and password.
IMPORTANT! You must create a separate Sample Entry Spreadsheet
for each 96-well plate.
How to Obtain More Information
Safety
Information
See the
GeneMapper
®
Software Version 4.1 Installation and
Administration Guide (PN 4403614)
for safety information.
Software
Warranty and
License
See the GeneMapper
®
Software Version 4.1 Installation and
Administration Guide (PN 4403614) for warranty and licensing
information.

Preface
How to Obtain More Information
GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide vii
Related
Documentation
The following related documents are shipped with the software:
• GeneMapper
®
Software Version 4.1 Installation and
Administration Guide (PN 4403614) – Provides procedures for
installing, securing, and maintaining version 4.1 of the
GeneMapper Software.
• GeneMapper
®
Software Version 4.1 Getting Started Guides for
microsatellite analysis (PN 4403672), loss of hetereozygosity
(LOH) analysis (PN 4403621), AFLP
®
system analysis
(PN 4403620), SNaPshot
®
kit analysis (PN 4403618), and
SNPlex
™
system analysis (PN 4403617) – Five guides that
explain how to analyze the application-specific example data
provided with the GeneMapper Software. The guides provide
brief, step-by-step procedures for the analysis of microsatellite,
LOH, AFLP
®
system, SNaPshot
®
kit, and SNPlex
™
system data
generated by compatible Applied Biosystems electrophoresis
instruments and Data Collection Software. The guides are
designed to help you quickly learn to use basic functions of the
GeneMapper Software.
• GeneMapper
®
Software Version 4.1 Online Help – Describes
the GeneMapper Software and provides procedures for common
tasks. Access online help by pressing F1, selecting Help
Contents and Index, or clicking in the toolbar of the
GeneMapper window.
• GeneMapper
®
Software Version 4.1 Quick Reference Guide
(PN 4403615) – Provides workflows for specific analysis types
and lists instruments, software, and analysis applications
compatible with the GeneMapper Software.
• GeneMapper
®
Software Version 4.1 Reference and
Troubleshooting Guide (PN 4403673) – Provides reference
information such as theory of operation and includes
troubleshooting information.
Portable document format (PDF) versions of this guide and the other
documents listed above are available on the GeneMapper
®
Software
Version 4.1 Documentation CD.
Note: For additional documentation, see “How to Obtain Support”
on page viii.

Preface
How to Obtain Support
viii GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide
Send Us Your
Comments
Applied Biosystems welcomes your comments and suggestions for
improving its user documents. You can e-mail your comments to:
techpubs@appliedbiosystems.com
Obtaining
Information from
the Online Help
The GeneMapper
®
Software features an online help system that
describes how to use each feature of the user interface. To access the
online help, click in any window or dialog box (HelpContents
and Index if available) for more information.
How to Obtain Support
For the latest services and support information for all locations, go to
http://www.appliedbiosystems.com
, then click the link for
Support
.
At the Support page, you can:
• Search through frequently asked questions (FAQs)
• Submit a question directly to Technical Support
• Order Applied Biosystems user documents, MSDSs, certificates
of analysis, and other related documents
• Download PDF documents
• Obtain information about customer training
• Download software updates and patches
In addition, the Support page provides access to worldwide telephone
and fax numbers to contact Applied Biosystems Technical Support
and Sales facilities.

Chapter 1
GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide 1
Chapter 1
Process Quality Values
and
Basic Troubleshooting
C
ha
p
ter
3
Al
g
orithms
C
hapter 2
S
NPlex
™
Sy
stem
Troubleshootin
g
Process Quality Values and
Basic Troubleshooting
In this chapter:
■ Diagnosing and Resolving Basic Problems . . . . . . . . . . . . . . 2
■ ADO (Allele Display Overflow) . . . . . . . . . . . . . . . . . . . . . . 8
■ AE (Allele Edit) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 8
■ AN (Allele Number). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9
■ BD (Broad Peak) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9
■ BIN (Out of Bin Allele) . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10
■ CC (Control Concordance). . . . . . . . . . . . . . . . . . . . . . . . . . . 11
■ DP (Double Peak). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12
■ GQ (Genotype Quality) . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13
■ LPH (Low Peak Height). . . . . . . . . . . . . . . . . . . . . . . . . . . . 15
■ MNF (Matrix Not Found) . . . . . . . . . . . . . . . . . . . . . . . . . . 16
■ NB (Narrow Bin) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18
■ OBA (One Basepair Allele) . . . . . . . . . . . . . . . . . . . . . . . . . 18
■ OS (Offscale) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 19
■ OVL (Overlap) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20
■ PHR (Peak Height Ratio) . . . . . . . . . . . . . . . . . . . . . . . . . . . 21
■ SFNF (Sample File Not Found) . . . . . . . . . . . . . . . . . . . . . . 22
■ SHP (Sharp Peak). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23
■ SNF (Size Standard Not Found). . . . . . . . . . . . . . . . . . . . . . 23
■ SP (Split Peak) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24
■ SPA (Single Peak Artifact). . . . . . . . . . . . . . . . . . . . . . . . . . 24
■ SPU (Spectral Pull-Up) . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25
■ SQ (Sizing Quality) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25
■ XTLK (Cross Talk) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 32

Chapter 1 Process Quality Values and Basic Troubleshooting
Diagnosing and Resolving Basic Problems
2 GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide
Diagnosing and Resolving Basic Problems
PQVs and the
Troubleshooting
Process
The GeneMapper
®
Software has a system of Process Quality Values
(PQVs) that provide the basis for troubleshooting fragment analysis
data using the software. PQVs are application-specific metrics, where
each evaluates the data for a specific quality that is consistent with a
problem associated with the type of analysis. In this way, the PQV
system can alert you to potential problems and provide you with a
starting point for investigation.
About Process
Quality Values
(PQVs)
Each individual PQV displays the result of a unique algorithmic test
that evaluates a specific property of the fragment analysis data. The
software performs the PQV tests in a specific sequence during the
analysis. With the exception of the Sizing Quality (SQ) PQV, the
software completes the analysis of each sample in a project even if a
sample fails one or more PQV tests.
The majority of PQV metrics yield numeric values between 0 and 1,
where 1 indicates that the related sample data or genotype completely
passed the associated test. Following the analysis, the software uses
the upper and lower thresholds for each PQV to translate the numeric
score into one of three symbols, displayed in the Samples or
Genotypes tab of the GeneMapper window.
Note:
If the thresholds of a PQV can be customized, the software
displays the parameters in the Quality Flags tab of the analysis method.
Note: Applied Biosystems recommends examining all samples that
produce (Check) or (Low Quality).
Symbol Definition
Default
Range
Pass: The sample or genotype passed the PQV
test.
0.75 to 1.0
Check: A possible problem exists for the sample
or genotype.
0.25 to 0.75
Low Quality/Fail: There is a strong possibility that
a problem exists for the sample or genotype.
0.0 to 0.25

Chapter 1 Process Quality Values and Basic Troubleshooting
Diagnosing and Resolving Basic Problems
GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide 3
Note: The Allele Display Overflow (ADO) and Allele Edit (AE)
PQVs of the Genotypes tab report their results as instead of the
colored icons.
Note:
The GeneMapper Software does not complete the analysis of
samples that display (Low Quality) for the Sizing Quality (SQ)
PQV test.
Adjusting the Threshold Settings of PQV Tests
Some PQV metrics include components that can be customized. In
those cases, the user-defined parameters for the PQV appear in the
Peak Quality tab of analysis methods for the applicable analysis type.
Adjusting the Weights of PQV
The majority of PQV contribute to the Genotype Quality (GQ) PQV, a
metric used to gauge the confidence of each genotype call. In those
cases, some PQV contain user-defined weights that determine how
significantly the associated PQV affect the GQ PQV calculation. For
those PQV, the user-defined weights appear in the Quality Flag tab of
analysis methods for the applicable analysis type. For more information
on the calculation of the GQ PQV, see “GQ (Genotype Quality)” on
page 13.
Note:
You can configure a PQV so that it does not contribute to the
GQ (by setting the weight to 0). However, the PQV remains active.
Rules for PQV Columns
• Quality metrics with Pass/ Check values and no Low
Quality values are warning flags. Analysis does not stop if problems
are detected with these properties, but Applied Biosystems
recommends examining results flagged as (Check).
• Holding the cursor over a column header displays a tooltip that lists
the full name of the column (the default names are often acronyms).

Chapter 1 Process Quality Values and Basic Troubleshooting
Diagnosing and Resolving Basic Problems
4 GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide
Tabl e 1-1 PQV values by application
PQVs
Analysis
See Page
AFLP
®
System
Microsatelite
SNaPshot
®
Kit
SNPlex
™
System
Abb. Name
2n
Other
ADO Allele Display Overflow
✔ ✔ ✔ ✔ ✔
8
AE Allele Edit
✔ ✔ ✔ ✔ ✔
8
AN Allele Number
✔ ✔ ✔ ✔
9
BD Broad Peak
✔ ✔ ✔ ✔ ✔
9
BIN Out of Bin Allele
✔ ✔ ✔
10
CC Control Concordance
✔ ✔ ✔ ✔
11
DP Double Peak
✔
12
GQ Genotype Quality
✔ ✔ ✔ ✔ ✔
13
LPH Low Peak Height
✔ ✔ ✔ ✔
15
MNF Matrix Not Found
✔ ✔ ✔ ✔
16
NB Narrow Bin
✔
18
OBA One Basepair Allele
✔
18
OS Off-Scale
✔ ✔ ✔ ✔ ✔
19
PHR Peak Height Ratio
✔ ✔ ✔ ✔
21
SFNF Sample File Not Found
✔ ✔ ✔ ✔ ✔
22
SHP Sharp Peak
✔
23
SNF Size Standard Not Found
✔ ✔ ✔ ✔ ✔
23
SP Split Peak
✔
24
SPA Single Peak Artifact
✔
24
SPU Spectral Pull-Up
✔ ✔ ✔ ✔ ✔
25
SQ Sizing Quality
✔ ✔ ✔ ✔ ✔
25
XTLK Cross Talk
✔ ✔ ✔
8

Chapter 1 Process Quality Values and Basic Troubleshooting
Diagnosing and Resolving Basic Problems
GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide 5
Common Troubleshooting Procedures
About the
Procedures
Procedures commonly used to troubleshoot errors and irregularities
in fragment analysis data:
■ Displaying Sample Information/Raw and EPT Data . see below
■ Displaying Numeric PQV Metrics . . . . . . . . . . . . . . . . . . . . . . 7
Displaying Sample
Information/Raw
and EPT Data
1. In the GeneMapper window, select the Samples tab.
2. In the Navigation Pane of the GeneMapper window:
a. Click to expand the contents of the project folder.
b. From the list of samples, select a sample that displayed
(Check) or (Low Quality).
3. Select the Info, Raw Data, and EPT Data tabs as needed to
display the sample information for the selected file.
• Info tab – Displays a summary of all information for the
associated sample file (see Table 1-2 on page 6).
•
Raw Data tab
– Displays an electropherogram of spectral
data collected during the run of the associated sample. The
spectral data is displayed in relative fluorescent units (RFU).
• EPT Data tab – Displays the EPT (electrical, power, and
temperature) data for the associated sample throughout the
course of the run.
Note: Table 1-2 lists only the information contained in the Info tab
that is relevant to troubleshooting. For a complete description of the
elements listed in the Info tab, see the GeneMapper
®
Software Online
Help.

Chapter 1 Process Quality Values and Basic Troubleshooting
Diagnosing and Resolving Basic Problems
6 GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide
Table 1-2 Info Tab information relevant to troubleshooting
Group
Description/Information
Sample
Information
Describes the source and status of the imported sample data.
Sample Origin Path – Displays the path to the associated sample file at the time it
was imported (provided that the sample was added to the project from a sample
file).
Status Message – Displays information related to any events that occurred when
the sample was imported into the project.
Error Message
Displays any errors the software encounters during the analysis of the associated
sample. You can use the information in this group to verify the source of several
analysis problems.
Current Settings
Describes the analysis settings currently applied to the associated sample.
All of the data displayed in this group is useful for troubleshooting problems with
the GeneMapper Software. Although, most of the information can be viewed in
various parts of the software, the Current Settings group summarizes all the
information in a single location for easier access.
Run Information
Provides basic information about the configuration of the compatible
Applied Biosystems electrophoresis instrument and the run itself.
Instrument Name – Displays the name of the instrument used to run the sample.
The instrument name is important when diagnosing trends in analysis errors that
can be traced back to the instrument used to run the failed sample(s).
Data Collection Ver – Displays the version number of the Data Collection
Software used to run the sample.
Data Collection
Settings
Describes the configuration of the Data Collection Software at the time the sample
was run.
The data displayed in this group can be used to troubleshoot problems caused by
modifications to run modules. By comparing the
Data Collection Settings
information from passing and failing runs, you can identify any changes made to
the run module (intentional or unintentional) that may have caused or contributed
to the failure.
Note: The data displayed in the EPT tab provides a log of the actual parameters
throughout the run.
Capillary
Information
Provides the basic specifications of the capillary array used to run the sample.
Capillary Number – Displays the number of the capillary used to run the
associated sample. Like the instrument name, the capillary number is important
when diagnosing trends in analysis errors that can be traced back to the capillary
used to run the failed sample(s).
Chapter 1 Process Quality Values and Basic Troubleshooting
Diagnosing and Resolving Basic Problems
GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide 7
Displaying
Numeric PQV
Metrics
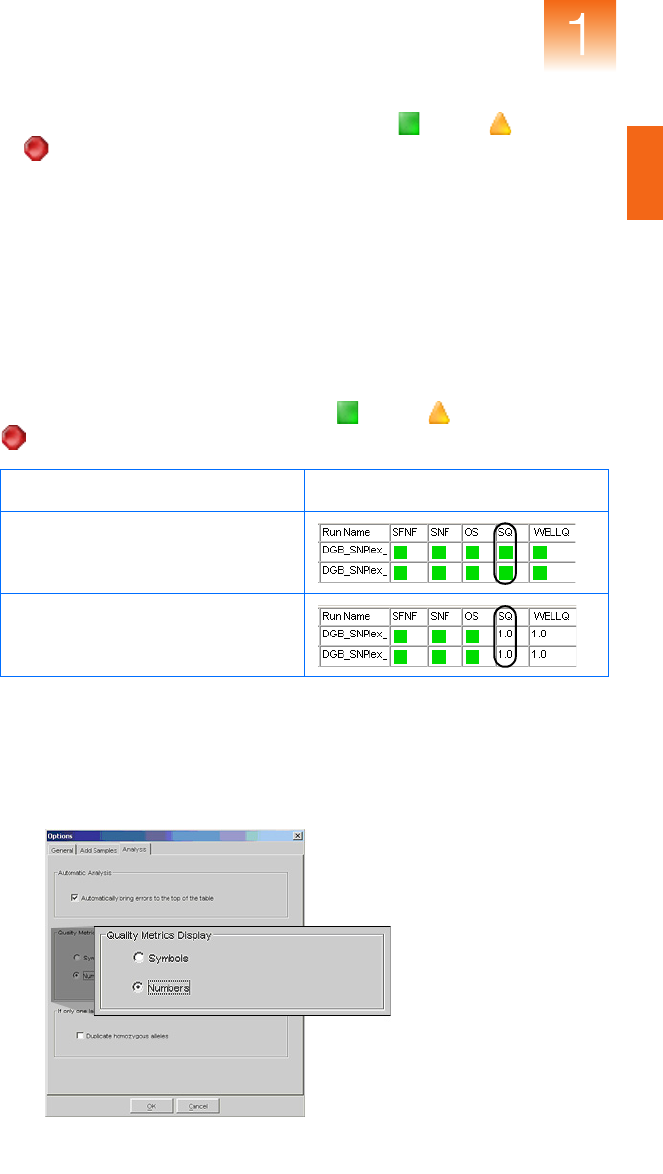
By default, the GeneMapper Software displays (Pass), (Check),
or (Low Quality) in some PQV columns to represent the numeric
score of the associated quality metric. When troubleshooting quality
errors, it is often more useful to configure the software to display
numeric representations of the quality values.
For example, the Sizing Quality (SQ) PQV evaluates the similarity
between the fragment pattern defined by the size standard definition and
the actual size standard peak distribution pattern in the sample data. The
Sizing Quality metric yields a value between 1 and 0 that represents a
combination of statistical measures for the size calling method used to
perform the analysis. Based on the PQV Threshold settings of the
Quality Flags tab, the software displays (Pass), (Check), or
(Low Quality) to indicate the result of the Sizing Quality calculation.
To display numerical representations of the quality metrics:
1. Select ToolsOptions, then select the Analysis tab.
2. In the Quality Metrics Display settings, select Numbers.
3. Click OK to apply the settings.
Sizing Quality Representation Samples Table Example
Symbols
(default)
Numbers
(recommended for troubleshooting)

Chapter 1 Process Quality Values and Basic Troubleshooting
ADO (Allele Display Overflow)
8 GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide
ADO (Allele Display Overflow)
Description/
Function
The ADO PQV indicates that the number of alleles called for the
associated sample at the specified marker exceeds the Allele Setting in
the Genoptypes tab of the table setting. Because the software is
configured to display fewer alleles than are present, the data for the
additional allele is hidden from view.
Note:
For each allele detected by the software, the Genotypes tab
displays six columns: name, size, height, area, mutation, and comments.
Expected Values – Indicates that the associated sample contains a number of
alleles at the specified marker that is greater than the user-defined
limit.
Troubleshooting Select the affected genotype, click (AnalysisDisplay Plots),
then review the affected sample for miscalled peaks.
AE (Allele Edit)
Description/
Function
The
AE PQV
indicates whether or not a user modified the allele call
for the associated genotype.
Note: Allele calls can be modified in the Samples Plot, the
Genotypes Plot, and the Cluster Plot.
Expected Values – Indicates that the associated genotype call has been edited.

Chapter 1 Process Quality Values and Basic Troubleshooting
AN (Allele Number)
GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide 9
AN (Allele Number)
Description/
Function
The AN PQV indicates that the associated sample contains either:
• A number of alleles at the specified marker that exceeds the
Max Expected Alleles setting (in the Peak Quality tab of the
analysis method)
or
• No alleles are present at the specified marker
Expected Values (Pass) or (Check)
Troubleshooting
Select the affected genotype(s), click (
Analysis
Display Plots
),
then review the sample data at the affected marker for additional peaks
or for the absence of peaks.
BD (Broad Peak)
Description/
Function
The BD PQV indicates that the width of the peak for the associated
genotype exceeds the Max peak width setting (in the Peak Quality tab of
the analysis method).
Note: When the BD PQV is triggered, the software reduces the GQ
PQV by 50% because the default multiplier is 0.5.
Expected Values (Pass) or (Check)
Troubleshooting Select the affected genotype, click (AnalysisDisplay Plots),
then review the associated peak for irregularities.

Chapter 1 Process Quality Values and Basic Troubleshooting
BIN (Out of Bin Allele)
10 GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide
BIN (Out of Bin Allele)
Description/
Function
The BIN PQV indicates that the apex of the peak for the associated
genotype is outside of the boundaries that define the associated bin.
Note: When the BIN PQV is triggered, the software reduces the GQ
PQV by 80% because the default multiplier is 0.8.
Note:
For human identification (HID) analysis, the BIN PQV is
displayed as the OL (Off-Ladder Alleles) PQV.
Expected Values (Pass) or (Check)
Troubleshooting Select the affected genotype, click (AnalysisDisplay Plots),
then review the allele(s) at the appropriate bin location.
Symptom Possible Cause Solution
BIN PQV displays
(Check)
After using Auto Bin to generate bins,
the software did not create a bin for an
allele peak because it considered the
peak to be a single peak artifact. The
SPA flag was triggered because the
software did not detect any stutter
peaks to the left of the allele peak; a
result of the minimum fragment length
for the marker being set too high
Correct the SPA flag by editing the
marker minimum fragment length,
then reanalyze and perform the Auto
Bin again.
The GeneMapper Software detected
an allele peak that did not fit into any
of the defined bins because the bins
were not calibrated to the allelic
ladder; a result of a sample file
containing an allelic ladder that is not
designated as an allelic ladder in the
Samples tab.
In the Samples tab of the
GeneMapper window, set the Sample
Type of the sample containing the
allelic ladder to
Allelic Ladder
.
You generated bins using the Auto Bin
function but the GQ value for a marker
was less than the Minimum Quality
Value of 0.1 (as set in the Auto Bin
dialog box).
View the allele peak(s) for the marker
in the Genotypes Plot window.
Determine if the allele peaks(s) are
valid. If so, manually create bin(s) for
the peak(s).

Chapter 1 Process Quality Values and Basic Troubleshooting
CC (Control Concordance)
GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide 11
CC (Control Concordance)
Description/
Function
The CC PQV indicates that the associated control sample does not
exactly match the defined alleles for the related marker.
IMPORTANT! Applied Biosystems recommends running the control
sample at least once for every panel.
Note: The CC PQV serves primarily as an internal control for quality
assurance.
Expected Values (Pass) or (Check)
Troubleshooting
Select the affected control sample in the Samples tab of the GeneMapper
window, click (
Analysis
Display Plots
), then review the positions
of the peaks relative to the bins.
Symptom Possible Cause Solution
CC PQV displays
(Check)
The allele calls of the sample defined
as the Positive Control in the Samples
tab do not match the Positive Control
allele calls in the marker definition
because the well contains the
incorrect positive control sample.
Run the correct positive control and
add the sample file to the project,
then define the sample as the
Positive Control in the Samples tab
of the GeneMapper window.
The allele calls of the sample defined
as the Positive Control in the Samples
tab do not match the positive control
allele calls in the marker definition
because the alleles were defined
incorrectly.
Edit the Positive Control allele calls in
the marker definition in the Panel
Manager.
The sample defined as the Negative
Control contains an “allele peak” due
to the presence of a spike caused by
dust or a gas bubble.
Rerun the negative control and add
the sample file to the project, then
define the sample as the Negative
Control in the Samples tab of the
GeneMapper window.

Chapter 1 Process Quality Values and Basic Troubleshooting
DP (Double Peak)
12 GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide
DP (Double Peak)
Description/
Function
The DP PQV indicates that the peak for the associated genotype:
• Resides in a bin with another peak of the same dye color
and
• The ratio of the peak height (minor/major peak height) is greater
than the Double Peak setting in the Peak Quality tab of the
Analysis Method
Expected Values (Pass) or (Check)
Troubleshooting
Select the affected genotype, click (
Analysis
Display Plots
), then
review the sample data at the appropriate bin for additional peaks
.
Symptom Possible Cause Solution
DP PQV displays
(Check)
A problem with the chemistry is
causing peaks from two different
markers not to resolve, possibly
because either of the primers are too
similar in length or the mobilities of
the two primer fragments are similar.
Check primer lengths and
electrophoresis conditions and
adjust as necessary.

Chapter 1 Process Quality Values and Basic Troubleshooting
GQ (Genotype Quality)
GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide 13
GQ (Genotype Quality)
Description/
Function
The GQ PQV provides a summary of the quality metrics for each
genotype. The GQ value is a calculated combination of the relevant,
weighted PQVs and the Marker Quality value for the genotype.
Calculation of the Genotype Quality (GQ) Metric
The formulas used by the GeneMapper Software to calculate the GQ
value are analysis-specific, and differ largely based on the PQVs
supported by each application. The following general formula
describes the genotype quality calculation:
where the Marker Quality (MQ) value is modified by the user-defined
PQVs to generate the final GQ value, and the PQVs are weighted from 0
to 1. The actual value of each PQV in the equation is 1 minus the weight
assigned in the Quality Flags tab of the analysis method used to analyze
the data.
IMPORTANT! The filtering of individual PQVs is controlled by the
threshold settings in the Peak Quality tab of the analysis method.
Also, the PQVs remain fully functional regardless of the weights
used.
PQV Weight Net Effect on GQ Calculation
0
No effect on the GQ calculation
The initial value of 1 minus the weight of 0 yields a PQV of
1. When used in the GQ calculation, the PQV has no effect
since 1
∞
MQ = MQ.
1
Reduces the GQ value to 0
The initial value of 1 minus the weight of 1 yields a PQV of
0. When used in the GQ calculation, the PQV automatically
causes the GQ to fail since 0
∞
MQ = 0.
0<x <1
Reduces the GQ value to the fraction specified by the
weight. The higher the value, the greater the effect on GQ.
GQ MQ 1 BD–()1OS–()…1SPU–()×××()×=

Chapter 1 Process Quality Values and Basic Troubleshooting
GQ (Genotype Quality)
14 GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide
Calculation of the Marker Quality Metric
Figure 1-1 shows how the GeneMapper Software generates a Marker
Quality (MQ) value from sample peak data with assigned Allele
Quality (AQ) values. AQ values are a function of quality value
assignments for: sizing quality, allele calling quality, bin assignment
quality, and bin quality.
Note:
When analyzing SNPlex
™
System sample data, the GeneMapper
Software calculates GQ values depending on the method (Model or
Rules) selected to perform allele calling. The following figure
illustrates the derivation of GQ values using the Rules method.
Figure 1-1 Calculation of the Marker Quality metric
Expected Values (Pass), (Check), or (Fail)
Note: The software assigns the GQ PQV flags based on the PQV
threshold settings in the Quality Flags tab of the analysis method.
Troubleshooting
Review the PQV for the affected genotype to determine the metric that
is causing the GQ PQV to fail.
Note:
To better determine how individual PQV contribute to the GQ
PQV, configure the software to display the PQV numerically, as
explained in “Displaying Numeric PQV Metrics” on page 7.
AQ
x (AQ) = MQ
AQ
GR2119
AQ
AQ

Chapter 1 Process Quality Values and Basic Troubleshooting
LPH (Low Peak Height)
GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide 15
LPH (Low Peak Height)
Description/
Function
The LPH PQV indicates that the height of the peak for the associated
genotype is lower than the associated heterozygous or homozygous
height limit that is specified in the analysis method. You can set
homozygous value (default is 200) and heterozygous value (default is
100) in the Peak Quality tab of the analysis method.
Note: When the LPH PQV is triggered, the software reduces the GQ
PQV by 50% (the default multiplier is “0.5”).
Expected Values (Pass) or (Check)
Troubleshooting Select the affected genotype, click (AnalysisDisplay Plots),
then review the associated peak for irregularities.

Chapter 1 Process Quality Values and Basic Troubleshooting
MNF (Matrix Not Found)
16 GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide
MNF (Matrix Not Found)
Description/
Function
The MNF PQV indicates whether or not the GeneMapper Software
can access the matrix specified in the Matrix column of the Samples
tab for the associated sample.
IMPORTANT! Because recent models of Applied Biosystems
instruments save matrix data to the sample files they create, the MNF
flag is applicable only to sample files created by the ABI P
RISM
®
310
and 377 instruments.
Expected Values (Pass) or (Check)
Troubleshooting Determine the name of the missing matrix file(s) by reviewing the
sample information for the affected samples, as explained in
“Displaying Sample Information/Raw and EPT Data” on page 5.
Importing a
Matrix File
(Windows
®
Only)
IMPORTANT!
You must convert matrix files created by Macintosh
®
computers before importing them. The conversion utility is free and
available from the software support section of the Applied Biosystems
website (
www.appliedbiosystems.com/support/software/
).
1. Click (ToolsGeneMapper Manager).
2. In the GeneMapper Manager, select the Matrices tab, then
click Import.
3. In the Importing Matrix dialog box, navigate to and select the
matrix file, then click Import.
4. Click Done to close the GeneMapper Manager, then analyze the
project.
Symptom Possible Cause Solution
MNF PQV displays
(Check)
The software could not
access the matrix file
specified in the Matrix
column for the associated
sample file.
Locate the missing matrix file, then import it as
explained in “Importing a Matrix File (Windows®
Only)” on page 16
If the sample files for the matrix standards used
to create the missing matrix are available, re-
create the matrix as explained in “Generating a
Matrix” on page 17.
Chapter 1 Process Quality Values and Basic Troubleshooting
MNF (Matrix Not Found)
GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide 17
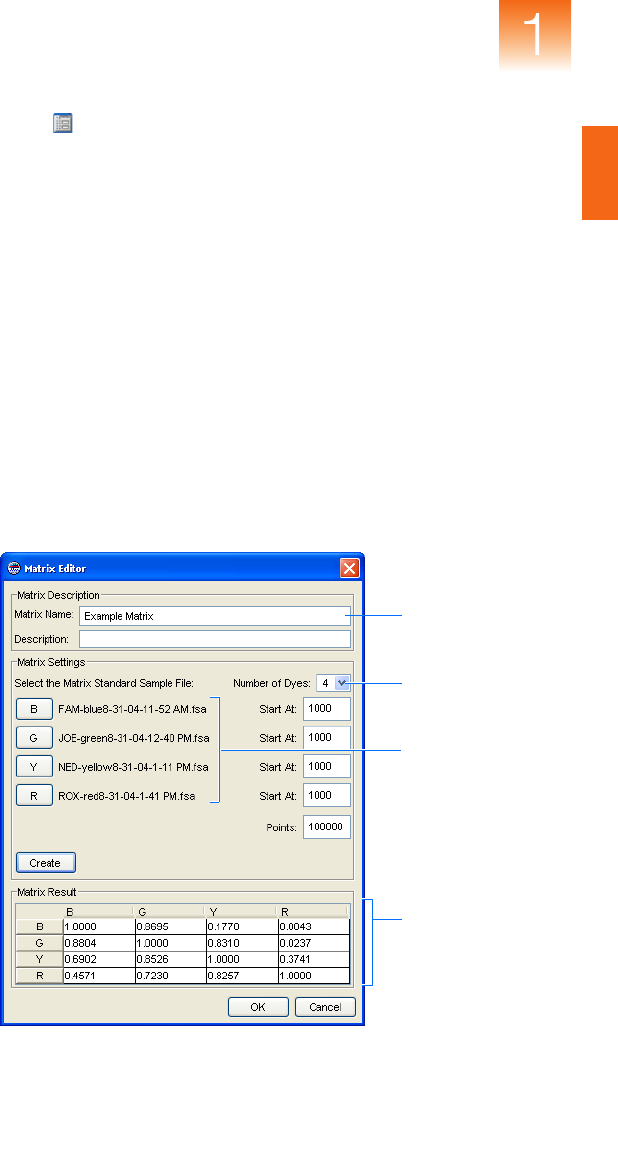
Generating a
Matrix
1. Click (ToolsGeneMapper Manager).
2. In the GeneMapper Manager, select the Matrices tab, then
click New.
3. In the Matrix Editor dialog box:
a. Type a name and description for the matrix.
b. In the Number of Dyes drop-down list, select the number of
dyes present in the matrix (4 or 5).
c. Click B, navigate to and select the sample file for the blue
matrix standard, then click Open.
d. Repeat step 3c for the remaining dyes in the matrix (Green,
Yellow, Red, and Orange if applicable).
e. Click Create to create the matrix.
f. Click OK.
4. Click Done to close the GeneMapper Manager.
5. In the Matrix column of the Samples tab, select the new matrix,
then analyze the project.
Size standard
name
Fragment sizes
for the size
standard
Size standard dye
channel
Generated matrix
data

Chapter 1 Process Quality Values and Basic Troubleshooting
NB (Narrow Bin)
18 GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide
NB (Narrow Bin)
Description/
Function
The NB PQV indicates that the apex of the associated peak for the
associated genotype is present within 0.5 base pairs of a bin that does
not contain a peak. This PQV is designed to capture peaks that are
outside of bin boundaries because of incorrect bin definitions.
Expected Values (Pass) or (Check)
Troubleshooting Select the affected genotype, click (AnalysisDisplay Plots),
then review the peak at the appropriate bin location.
OBA (One Basepair Allele)
Description/
Function
The OBA PQV indicates, for the associated genotype, that the apex
of the associated peak is present at a position within 1 base pair of
another peak.
Expected Values (Pass) or (Check)
Troubleshooting Select the affected genotype, click (AnalysisDisplay Plots),
then review the allele at the appropriate bin location for a
microvariant peak or an invalid allele call.
Symptom Possible Cause Solution
NB PQV displays
(Check)
You created a bin that is too narrow
to contain its associated allele peak.
In the Panel Manager, edit the bin
width and/or location so that it
contains the allele peak.

Chapter 1 Process Quality Values and Basic Troubleshooting
OS (Offscale)
GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide 19
OS (Offscale)
Description/
Function
The OS PQV is displayed in both the Samples and Genotypes tabs of
the GeneMapper window, but the function of the OS PQV in each tab
differs in the following way:
• OS PQV for the Samples tab – The signal associated with the
size standard of the specified sample contains one or more peaks
that exceed the maximum detectable range.
• OS PQV for the Genotypes Tab – The signals associated with
the given sample contain one or more peaks that exceed the
maximum detectable range.
Note:
When the OS PQV of the Genotypes tab is triggered, the
software reduces the GQ PQV by 50% (the default multiplier is “0.5”).
Expected Values (Pass) or (Check)
Troubleshooting 1.
In the GeneMapper window, select the
Samples
tab.
2.
In the Navigation Pane, click to expand the project folder, then
select the sample that displays (Check) in the OS column.
3.
Select the
Raw Data
tab to display the electropherogram of
normalized spectral data collected during the associated sample run.
The spectral data is displayed in Relative Fluorescent Units (RFU).
4.
Review the data for offscale peaks.
5. Use Table 1-3 on page 20 to determine an appropriate corrective
action.
Offscale peak
(primer peak)

Chapter 1 Process Quality Values and Basic Troubleshooting
OVL (Overlap)
20 GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide
OVL (Overlap)
Description/
Function
The OVL PQV indicates that the peak for the associated genotype
has been called twice by the GeneMapper Software. If the ranges of
two bins overlap, a peak can reside in both bins and, therefore, be
called twice, once for each allele.
Expected Values (Pass) or (Check)
Troubleshooting Select the affected genotype, click (AnalysisDisplay Plots),
then review the peak and associated bins at the appropriate location.
Table 1-3 OS PQV Troubleshooting
Symptom Possible Cause Solution
• MNF PQV in the Samples tab displays
(Check)
• Raw data contains multiple off-scale
size standard peaks
Too much size
standard injected
into the capillary
No action necessary. The data
cannot be manipulated to remove
the oversized peaks.
Decrease the quantity of size
standard used in subsequent runs.
Also, make sure to use Hi-Di
Formamide as the loading reagent.
IMPORTANT! Water loading can
produce artificially high signal and
is not recommended.
• MNF PQV in the Genotypes tab
displays (Check)
• Raw data contains multiple off-scale
peaks in the signal(s) associated with
the sample fragments
Too much sample
injected into the
capillary
No action necessary. The data
cannot be manipulated to remove
the oversized peaks.
Decrease the quantity of sample
used in subsequent runs.

Chapter 1 Process Quality Values and Basic Troubleshooting
PHR (Peak Height Ratio)
GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide 21
PHR (Peak Height Ratio)
Description/
Function
The PHR PQV indicates that the apex of the peak for the associated
genotype is:
• Present at a position within 1 base pair of another peak
and
• The ratio of the height of the lower peak to that of the higher
peak is less than the Minimum Peak Height Ratio setting in the
Peak Quality tab of the analysis method.
Note: For LMS markers, the ratio is calculated based on the peak
heights of the called allele peaks.
Note: For SNaPshot
®
kit analysis, the ratios are calculated as they
are for microsatellite markers except that they span two different
colors, and only two peaks are used in the calculation.
Expected Values (Pass) or (Check)
Troubleshooting Select the affected genotype, click (AnalysisDisplay Plots),
then review the peaks at the appropriate location.
Symptom Possible Cause Solution
PHR PQV displays
(Check)
The sample has undergone Loss of
Heterozygosity (LOH). A difference
in peak heights between alleles is
expected.
Normal occurrence. No action
necessary.
Further evaluate the sample for LOH
using the Report Settings Editor and
Report Manager.
For more information, see the
GeneMapper
®
Software Version 4.1
LOH Analysis Getting Started Guide
(PN 4403621).

Chapter 1 Process Quality Values and Basic Troubleshooting
SFNF (Sample File Not Found)
22 GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide
SFNF (Sample File Not Found)
Description/
Function
The SFNF PQV indicates whether or not the software can access the
sample file (*.fsa) shown in the Sample File column of the associated
sample. When the software adds a sample to a project from a sample file,
it retains a link to the original file. The software displays (Check) in
the SFNF column if the sample file is deleted, renamed, or moved.
Expected Values (Pass) or (Check)
Troubleshooting Determine the name and location of the missing sample file:
1. In the GeneMapper window, select the Samples tab.
2. In the Navigation Pane of the GeneMapper window, click to
expand the contents of the project folder, then select the sample
that display (Check) in the SFNF column.
3. Select the Info tab, then note the name (Sample File) and
location (Sample Origin Path) of the sample.
Symptom Possible Cause Solution
SFNF PQV displays
(Check)
Sample file has been
renamed, moved, or
deleted.
Search the local drives of the computer for the
sample file, then do one of the following:
• If you cannot find the file, no further action can
be taken to resolve the PQV flag.
• If you find the file, use the Associate Sample
feature to direct the software to the new
location as follows:
a. In the Samples tab of the GeneMapper
window, select the affected samples.
b. Select FileAssociate Samples.
c. In the Select Folder dialog box, select the
folder containing the missing files, then
click Select.

Chapter 1 Process Quality Values and Basic Troubleshooting
SHP (Sharp Peak)
GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide 23
SHP (Sharp Peak)
Description/
Function
The
SHP
PQV indicates
that the peak for the associated genotype is
part of a cluster of peaks with a large, narrow peak in the middle
whose width is 50% less than either of the neighboring peaks.
Expected Values (Pass) or (Check)
Troubleshooting Select the affected genotype, click (AnalysisDisplay Plots),
then review the peak at the appropriate location.
SNF (Size Standard Not Found)
Description/
Function
The
SNF PQV
indicates whether or not the GeneMapper Software
can access the
size standard definition
specified in the Size Standard
column for the associated sample.
Expected Values (Pass) or (Check)
Troubleshooting Verify that the software does not contain the desired size standard:
1. In the Samples tab of the GeneMapper window, note the name
of the size standard assigned to the affected sample.
2. Click (ToolsGeneMapper Manager).
3. In the GeneMapper Manager, select the Size Standards tab.
4. Verify that the Size Standard tab does not list the missing size
standard, or that it has not been renamed.
Symptom Possible Cause Solution
SNF PQV displays
(Check)
(Autoanalysis only) The size
standard may have been set
incorrectly in the plate record of the
Data Collection Software.
Do one of the following:
• If using an Applied Biosystems
size standard, click Import to
import the definition from the
default Panels folder.
•Click New to create a custom
size standard of the same name.
The size standard definition has been
renamed, deleted, or does not exist.

Chapter 1 Process Quality Values and Basic Troubleshooting
SP (Split Peak)
24 GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide
SP (Split Peak)
Description/
Function
The
SP PQV
indicates
that the peak for the associated genotype is
part of a pair of overlapping peaks that are less than 0.25 base pairs
apart (the horizontal distance between two peak apexes).
Expected Values (Pass) or (Check)
Troubleshooting Select the affected genotype, click (AnalysisDisplay Plots),
then review the peak at the appropriate location.
SPA (Single Peak Artifact)
Description/
Function
The
SPA PQV
indicates
that no peaks are present within a two-base-
pair range before the peak for the associated genotype. This PQV is
designed to detect the absence of stutter peaks that accompany
microsatellite peaks.
Expected Values (Pass) or (Check)
Troubleshooting Select the affected genotype, click (AnalysisDisplay Plots),
then review the peak at the appropriate location.
Symptom Possible Cause Solution
SPA PQV displays
(Check)
The GeneMapper Software did not
detect any stutter peaks to the left of
the allele peak because the
minimum fragment length for the
marker was set too high.
In the Panel Manager, edit the
marker minimum fragment length,
then reanalyze.

Chapter 1 Process Quality Values and Basic Troubleshooting
SPU (Spectral Pull-Up)
GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide 25
SPU (Spectral Pull-Up)
Description/
Function
The SPU PQV indicates that the apex of the peak for the associated
genotype is at a position where the marker signal contains pull-up
peaks (also called bleed-through peaks). Pull-up peaks occur when
the peak height of the called allele is less than X% of the larger peak
that is within ±1 data point.
Expected Values (Pass) or (Check)
Troubleshooting Select the affected genotype, click (AnalysisDisplay Plots),
then review the peak at the appropriate location.
SQ (Sizing Quality)
Description/
Function
The SQ PQV reports the result of the Sizing Quality test, which gauges
the similarity between the fragment pattern defined by the size
standard definition, and the actual distribution of size standard peaks in
the sample data. The metric of the Sizing Quality test is a combination
of several values which measure the success of the algorithms that:
• Identify and eliminate primer peaks based on peak shape
• Perform size matching (ratio matching)
• Make a size-calling curve using the chosen sizing method
The Sizing Quality metric yields a value between 0 and 1. Based on the
PQV Threshold settings in the analysis method used to analyze the
data, the software translates the metric into the (Pass), (Check),
or (Low Quality) flags to indicate the result of the test.
Note: The GeneMapper Software does not complete the analysis of
samples that fail the Sizing Quality test (samples that display ).
Expected Values (Pass), (Check), or (
Low Quality
)
Note: When performing size calling using the Classic sizing method,
the software cannot determine Sizing Quality and, therefore, SQ is
always
(Check)
.

Chapter 1 Process Quality Values and Basic Troubleshooting
SQ (Sizing Quality)
26 GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide
Troubleshooting Review the data of the size standards that failed the SQ PQV:
1. In the Samples tab of the GeneMapper window, click
(AnalysisLow Quality to Top) to sort the data so that the
samples that produced errors appear at the top of the table.
2. In the Samples tab, select the
rows for the
sample(s) that display
(Check) or (Fail) in the SQ column.
3. Click (AnalysisSize Match Editor) to view the sizing
information for the selected sample(s).
4.
In the Navigation Pane of the Size Match Editor, select a sample
file
to display the sizing data for the associated sample.
5.
Review the data for the following qualities
:
• Signal Strength – The signal strength (peak height) of all
peaks must exceed the Peak Detection Threshold defined in
the analysis method used to analyze the data.
• Correct Size Calls/Labels – All peaks must be correctly
identified by the software. The labels above the peaks must
be in sequential order from left to right, least to greatest.
• Evenness of Signal Strength – All peaks should have
relatively uniform signal strengths.
• Sizing Quality – The sizing quality of each sample should
be within the passing range for your chemistry application.
Note: To magnify the plot of the Size Matches tab, drag the
mouse cursor ( ) across a region of the x- or y-axis.
6. Use Table 1-4 on page 27 to determine an appropriate corrective
action.
7. Repeat steps 4 through 6 for each sample file.

Chapter 1 Process Quality Values and Basic Troubleshooting
SQ (Sizing Quality)
GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide 27
Table 1-4 SQ PQV Troubleshooting
Symptom Possible Cause Solution
•SQ PQV displays or
• Size Match Editor does not
display peak data
The Size Standard Dye setting for
the size standard definition is not
set to the correct dye.
1. Verify that the correct size
standard definition is in use.
2. Open the size standard
definition and verify that the:
– Dye setting is set to the
correct dye
– Fragment sizes are correct
3. Modify the size standard
definition as necessary.
•SQ PQV displays or
• Peaks do not contain size
labels
The fragment sizes of the size
standard definition do not match
the positions of the detected
peaks.
•SQ PQV displays or
• One or more miscalled peaks
Peak detection threshold
associated with the size standard
is set too high or low.
Adjust the analysis method so
that the peak detection threshold
associated with the size standard
is greater than the height of the
miscalled peak. See “Adjusting
Peak Detection Thresholds” on
page 29 for more information.
•SQ PQV displays or
• Peaks are clear and
distinguishable, but have
low signal strength
Peak detection threshold
associated with the size standard
is set too high or low.
Electrophoresis or pipetting error
•SQ PQV displays or
• Size standard peaks occur
within a primer peak
Insufficient cleanup step Create and analyze the data
using a custom size standard
that does not include the
undetectable peak. See
“Correcting Miscalled Peaks” on
page 28 for more information.
•SQ PQV displays or
• Size standard peaks are clear
and distinguishable, but
consistently have low signal
strength
Incorrect concentration of size
standard in sample loading
reagent
Increase the concentration of
size standard added to
subsequent runs.
Incorrect injection settings Review the injection settings of
the run module for errors.
•SQ PQV displays or
• Peaks are clear and
distinguishable, but have
low signal strength
• Sizing failures occur in a
regular pattern (the same
wells fail repeatedly)
• Electrophoresis or pipetting
error
• Defective capillaries/arrays
See the user manual for your
Applied Biosystems
electrophoresis instrument for
information on troubleshooting
defective capillaries/arrays.
•SQ PQV displays or
• Size calling errors occur for
different samples on the same
capillary over multiple runs
Defective capillary

Chapter 1 Process Quality Values and Basic Troubleshooting
SQ (Sizing Quality)
28 GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide
Correcting
Miscalled Peaks
You can use the Size Match Editor to correct peaks that are miscalled
by the GeneMapper Software.
To correct a miscalled size standard:
1. In the Navigation Pane of the Size Match Editor, select the
sample file containing the miscalled peak.
2. Remove the label from the miscalled peak:
a. Select the peak with the label by clicking inside the body of
the peak.
b. Select EditDelete Size Label (or right-click the peak,
then select Delete).
3. Apply the label to the correct peak:
a. Select the correct peak.
b. Select EditAdd Size Label (or right-click the peak, then
select Add).
c. In the Select Size dialog box, double-click the label to
apply to the selected peak.
4. Click (ToolsCheck Sizing Quality) to verify that the
sample sizes correctly.
5. Click Apply to save the changes, then click OK.
IMPORTANT! You must click Apply to reanalyze the sample.
Note: Observe that the cell in the Status column for the sample
now displays (Analysis Required).
6. Reanalyze the sample using the new setting to verify that the
problem is resolved.

Chapter 1 Process Quality Values and Basic Troubleshooting
SQ (Sizing Quality)
GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide 29
Adjusting Peak
Detection
Thresholds
You can resolve a significant number of sizing failures by adjusting
the peak detection thresholds of the analysis method for a project.
The software identifies peaks that exceed the threshold for each
associated dye channel, but it cannot identify peaks that fall below it.
Samples that exhibit low signal intensity (low peak heights) can
occasionally fail sizing because one or more peaks fall below the
threshold defined in the analysis method. By lowering the threshold
of the appropriate dye channel, the software can call the peak(s)
correctly.
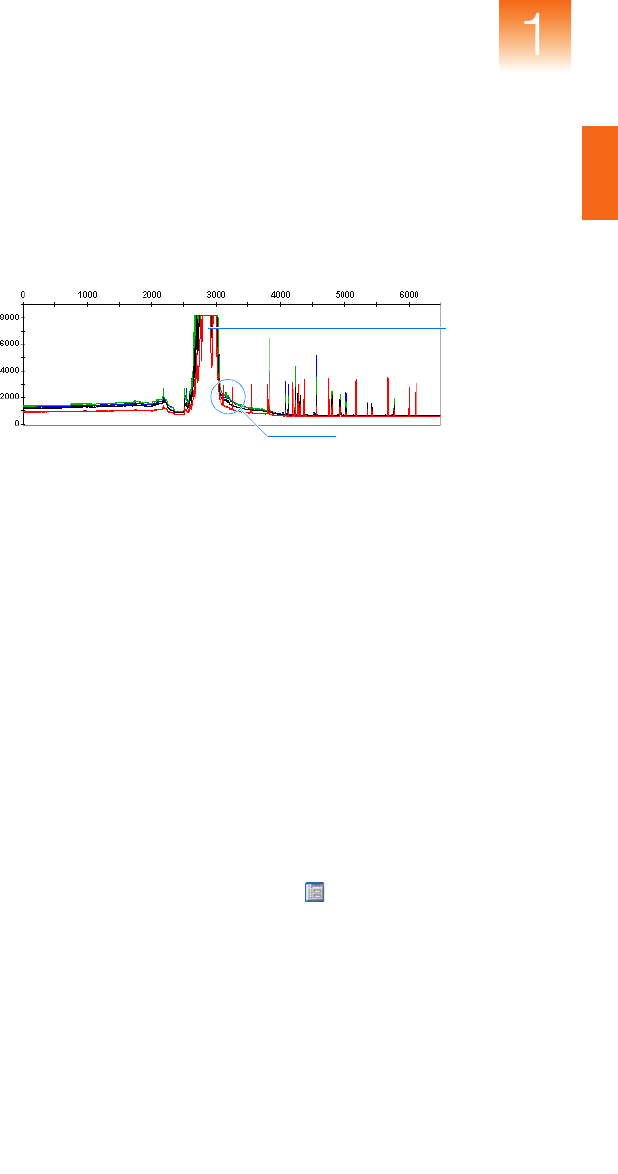
In contrast, when the signals of the size standard peaks are very high,
the software may misidentify a shoulder preceding a peak as the main
peak (see Figure 1-2). Because the shoulder peak does not occur at
the correct position relative to the other peaks, sizing fails. By
adjusting the analysis method so that the threshold value is greater
than the height of the shoulder, you can achieve good sizing.
Figure 1-2 Size standard with shoulder incorrectly labeled as a peak
To lower the peak detection thresholds of an analysis method:
1. In the GeneMapper window, click (ToolsGeneMapper
Manager).
2. In the GeneMapper Manager, select the Analysis Methods tab.
25-bp shoulder
incorrectly labeled
as a peak

Chapter 1 Process Quality Values and Basic Troubleshooting
SQ (Sizing Quality)
30 GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide
3. Do one of the following, to:
Modify the current analysis method, go to step 4.
Create a copy of the current analysis method:
a. Select the current analysis method.
b. Click Save As.
c. In the Save As dialog box, type a name for the new method,
then click OK.
4. Select the analysis method you want to modify, then click Open.
5. In the Analysis Method Editor, select the Peak Detector tab.
6. Modify the appropriate Peak Amplitude Threshold settings as
needed. Ideally, you should set the threshold of the appropriate
dye channel to a value less than the signal intensity of the
shortest size standard peak.
Note: Applied Biosystems recommends using Peak Amplitude
Threshold settings of no less than 50 RFU.
7. Click OK to save the analysis method.
8. Click Done to close the GeneMapper Manager, then reanalyze
the samples using the new analysis method.
Peak Amplitude
Threshold settings
Chapter 1 Process Quality Values and Basic Troubleshooting
SQ (Sizing Quality)
GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide 31
Customizing a
Size Standard
Definition
You can create a custom size standard definition to correct some
problems that consistently cause samples to fail sizing. Examples of
problems that you can resolve using custom size standards include
a
series of samples that fail sizing because
:
• The primer peak prevents the software from detecting and sizing
the peaks of the smaller size standard fragments
• A fragment of a custom size standard does not migrate as expected
during electrophoresis
About GeneMapper Software Size Standards
Before the GeneMapper Software can size fragment analysis data, it
must contain information about the size standard that was run with
the samples. The size standard definition supplies the software with
two pieces of information: the color of the dye associated with the
size standard, and the sizes (in bp) of the fragments that comprise the
size standard. Although the software provides definitions for all
Applied Biosystems size standards, you may need to create your own
definition if you choose to use a third-party standard, or experience
difficulty analyzing your data.
To create a custom size standard definition:
1. In the GeneMapper window, click (ToolsGeneMapper
Manager).
2. In the GeneMapper Manager, select the Size Standards tab.
3. Click New.
4. In the Select Dye and Analysis Method dialog box, select Basic
or Advanced, then click OK.
Size standard peaks
eclipsed by the primer peak
Primer peak

Chapter 1 Process Quality Values and Basic Troubleshooting
XTLK (Cross Talk)
32 GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide
5. In the Size Standard Editor:
a. In the Name field, type a name for the custom standard.
b. Select Size Standard Dye<appropriate dye>.
c. In the Size Standard table, type the size values for the size
standard (press Enter after typing each value).
IMPORTANT! After typing the last value, you must press Enter
to ensure the final value is included in the definition.
IMPORTANT! The values for the Analysis Range and Sizing
Range defined in the Allele and Peak Detector tabs of the
analysis method must match the peak range defined by the
associated size standard.
6. Click OK to save the size standard.
7. Click Done to close the GeneMapper Manager.
8. In the Samples tab of the GeneMapper window, apply the new
size standard to the samples of the project.
9. Reanalyze the sample using the new setting to verify that the
problem has been resolved.
XTLK (Cross Talk)
Description/
Function
The XTLK PQV indicates that, at the peak position of the associated
genotype, the ratio of the signals collected from the neighboring
capillaries exceed the Cross-talk ratio setting in the Peak Quality tab
of the analysis method.
Note: When the
XTLK
PQV is triggered, the software reduces the
GQ PQV by 50% (the default multiplier is “0.5”).
Expected Values (Pass) or (Check)

Chapter 2
GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide 33
Chapter 2
SNPlex™ System
Troubleshooting
C
ha
p
ter
3
Algorithm
s
C
ha
p
ter
1
P
rocess Quality Values
a
n
d
Basic
T
roubleshootin
g
SNPlex
™
System
Troubleshooting
In this chapter:
■ Overview . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 34

Chapter 2 SNPlex
™
System Troubleshooting
Overview
34 GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide
Overview
Identifying
Potential
Problems
You can use tools in the GeneMapper
®
Software to identify potential
problems at both the project and study levels of the SNPlex
™
System
analysis.
Identifying Potential Problems in SNPlex System Studies
Version 4.1 of the GeneMapper Software emphasizes the use of
studies for analyzing data generated using the SNPlex
™
Genotyping
System Chemistries. You can use the tools of the Study Manager to
maintain system-wide quality control and visualize potential
problems in SNPlex System data. After you identify a potential
problem, the software allows you to review the applicable run and
resolve the issues that can be corrected.
Note: See the GeneMapper
®
Software Version 4.1 SNPlex
™
System
Analysis Getting Started Guide (PN 4403617) for a detailed
explanation of the study management system.
Identifying Potential Problems in SNPlex Projects
SNPlex projects, like the other analyses supported by the
GeneMapper Software, contain a variety of Process Quality Values
(PQVs), which can aid you in identifying potential problems.
Chapter 1 explains the functions of all PQVs relevant to the analysis
of SNPlex System data.
Resolving
Problems and
Errors
The SNPlex
™
Genotyping System 48-Plex User Guide (PN 4360856)
describes how to resolve common chemistry- and software-related
problems. The user guide addresses all aspects of the SNPlex
System, not just those issues that are exclusive to the GeneMapper
Software analysis.

Chapter 3
GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide 35
Chapter 3
Algorithms
C
ha
p
ter
1
P
rocess Quality Values
a
n
d
Basic
T
roubleshootin
g
C
ha
p
ter 2
S
NPlex
™
System
T
rou
bl
es
h
oot
i
n
g
Algorithms
In this chapter:
■ Genotyping Algorithms . . . . . . . . . . . . . . . . . . . . . . . . . . . . 36
■ Peak Detection . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 38
■ Optimizing Peak Detection Sensitivity . . . . . . . . . . . . . . . . 41
■ Slope Thresholds for Peak Start/End Parameters. . . . . . . . . 44
■ Slope Threshold Example . . . . . . . . . . . . . . . . . . . . . . . . . . 45
■ Size-Matching/Size-Calling Algorithm . . . . . . . . . . . . . . . . 46
■ Size-Calling Methods (Classic and Advanced Modes) . . . . 47
■ Allele-Calling Algorithms . . . . . . . . . . . . . . . . . . . . . . . . . . 53
Chapter 3 Algorithms
Genotyping Algorithms
36 GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide
Genotyping Algorithms
Overview This chapter discusses the following algorithms:
• Peak Detection – Uses the Basic, Advanced, or Classic mode
algorithms to detect peaks and process data
•
Size-Matching/Calling
– Matches detected peaks to size standards
•
Binning
– Determines bin centers for genotyping
•
Allele-Calling
– Produces a consensus call based on several allele-
calling algorithms
Data Flow Figure 3-3 and Figure 3-4 on page 37 show the data flow in
GeneMapper
®
Software. Standard signal processing is applied to the
data before the data are delivered to the GeneMapper Software
algorithms.
Figure 3-3 Microsatellite analysis data flow
Input Sample
Supported Applied Biosystems
Genetic Analysis Systems
Baselining
Peak Detection
Binning
Size-Matching/Calling
Caller n
Arbitrator
Bin Assignment
Final Quality Value Determination
Report Results to User
Caller 1
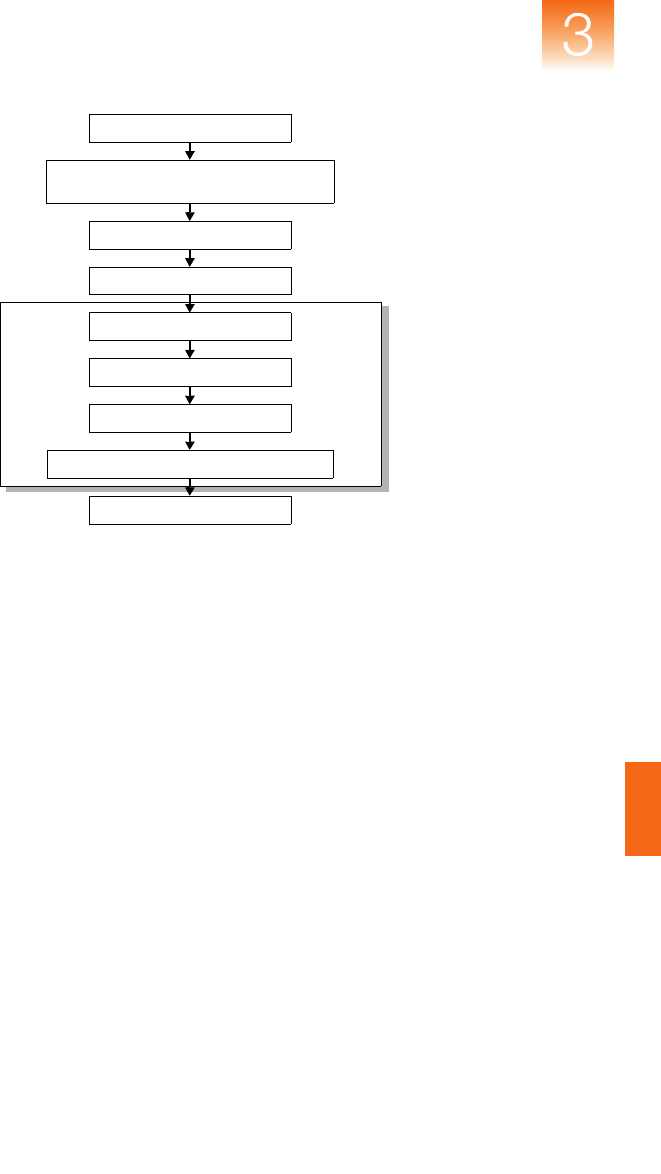
Chapter 3 Algorithms
Genotyping Algorithms
GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide 37
Figure 3-4 SNPlex
™
System analysis data flow
Input Sample
Supported Applied Biosystems
Genetic Analysis Systems
Baselining
Peak Detection
Binning
Size-Matching/Calling
Allele Calling
Final Quality Value Determination
Report Results to User

Chapter 3 Algorithms
Peak Detection
38 GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide
Peak Detection
Polynomial
Degree and Peak
Window Size
Parameters
Two peak-detection parameters are used in the polynomial detection
algorithm: Polynomial Degree, and Peak Window Size.
Use the Polynomial Degree and the Peak Window Size settings to
adjust the sensitivity of the peak detection. You can adjust these
parameters to detect a single base pair difference while minimizing
the detection of shoulder effects or noise.
Sensitivity increases with larger polynomial degree values and
smaller window size values. Conversely, sensitivity decreases with
smaller polynomial degree values and larger window size values.
How They Work
The peak window size functions with the polynomial degree to set
the sensitivity of peak detection. The peak detector calculates the first
derivative of a polynomial curve fitted to the data within a window
that is centered on each data point in the analysis range.
Using curves with larger polynomial degree values allows the curve
to more closely approximate the signal and, therefore, the peak
detector captures more peak structure in the electropherogram.
The peak window size sets the width (in data points) of the window
to which the polynomial curve is fitted to data. Higher peak window
size values smooth out the polynomial curve, which limits the
structure being detected. Smaller window size values allow a curve to
better fit the underlying data.
How to Use the Peak Detection Parameters
Use the table below to adjust the sensitivity of detection.
Function Polynomial Degree Value Window Size Value
Increase sensitivity Higher Lower
Decrease sensitivity Lower Higher

Chapter 3 Algorithms
Peak Detection
GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide 39
Guidelines for Use
To detect well-isolated, baseline-resolved peaks, use polynomial degree
values of 2 or 3. For finer control, use a degree value of 4 or greater.
As a guideline, set the peak window size (in data points) to be about 1
to 2 times the full width at half maximum height of the peaks that you
want to detect.
Examining Peak
Definitions
To examine how GeneMapper Software has defined a peak, select
ViewShow Peak Positions. The peak positions, including the
beginning, apex, and end of each peak, are tick-marked in the
electropherogram.
Effects of Varying
the Polynomial
Degree
Figure 3-5 shows peaks detected with a window size of 15 data
points and a polynomial curve of degree 2 (green), 3 (red), and 4
(black). The diamonds represent a detected peak using the respective
polynomial curves.
Note that the smaller trailing peak is not detected using a degree of 2
(green). As the peak detection window is applied to each data point
across the displayed region, a polynomial curve of degree 2 could not
be fitted to the underlying data to detect its structure.
Figure 3-5 Electropherogram showing peaks detected with three
different polynomial degrees
Polynomial curve of degree 4
(black)
Polynomial curve of degree 3
(red)
Polynomial curve of degree 2
(green)

Chapter 3 Algorithms
Peak Detection
40 GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide
Effects of
Increasing the
Window Size
Value
In Figure 3-6 both polynomial curves have a degree of 3 and the
window size value was increased from 15 (red) to 31(black) data
points.
As the cubic polynomial is stretched to fit the data in the larger
window size, the polynomial curve becomes smoother. Note that the
structure of the smaller trailing peak is no longer detected as a
distinct peak from the adjacent larger peak to the right.
Figure 3-6 Electropherogram showing the same peaks as in the
Figure 3-5 after increasing the window size value but keeping the
polynomial degree the same
Window size value of 15 (red)
Window size value of 31 (black)

Chapter 3 Algorithms
Optimizing Peak Detection Sensitivity
GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide 41
Optimizing Peak Detection Sensitivity
Examples ■ Example 1: Reducing Window Size. . . . . . . . . . . . . . . . . . . . 41
■ Example 2: Reducing Window Size/Increasing
Polynomial Degree. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 42
■ Example 3: Extreme Settings . . . . . . . . . . . . . . . . . . . . . . . . . 43
Example 1: Reducing Window Size
Initial
Electropherogram
Figure 3-7 shows two resolved alleles of known fragment lengths
(that differ by one nucleotide) detected as a single peak. The analysis
was performed using a polynomial degree of 3 and a peak window
size of 19 data points.
Figure 3-7 Electropherogram showing two resolved alleles
detected as a single peak
Note: For information on the tick marks displayed in the
electropherogram, see “Examining Peak Definitions” on page 39.

Chapter 3 Algorithms
Optimizing Peak Detection Sensitivity
42 GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide
Effects of
Reducing
Figure 3-8 shows that both alleles are detected after reanalyzing with
the polynomial degree set to 3 and the window size value decreased
to 15 (from 19) data points.
Figure 3-8 Electropherogram showing the alleles detected as
two peaks after decreasing the window size value
Example 2: Reducing Window Size/Increasing Polynomial Degree
Initial
Electropherogram
Figure 3-9 shows an analysis performed using a polynomial degree
of 3 and a peak window size of 19 data points.
Figure 3-9 Electropherogram showing four resolved peaks
detected as two peaks

Chapter 3 Algorithms
Optimizing Peak Detection Sensitivity
GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide 43
Effects of
Reducing the
Window Size Value
While Increasing
the Polynomial
Degree Value
Figure 3-10 shows the data presented in the figure above re-analyzed
with a window size value of 10 and polynomial degree value of 5.
Figure 3-10 Electropherogram showing all four peaks detected
after reducing the window size value and increasing the
polynomial degree value
Example 3: Extreme Settings
Effects of Extreme
Settings
Figure 3-11 shows the result of an analysis using a peak window size
value set to 10 and a polynomial degree set to 9. These extreme
settings for peak detection caused several peaks to be split and
detected as two separate peaks.
Figure 3-11 Electropherogram showing the result of an analysis
using extreme setting for peak detection

Chapter 3 Algorithms
Slope Thresholds for Peak Start/End Parameters
44 GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide
Slope Thresholds for Peak Start/End Parameters
About These
Parameters
Use the Slope Threshold for Peak Start and Slope Threshold for Peak
End parameters to adjust the start and end points of a peak.
The values assigned to these parameters can be used to better position
the start and end points of an asymmetrical peak, or a poorly resolved
shouldering peak to more accurately reflect the peak position and area.
How These
Parameters Work
In general, from left to right, the slope of a peak increases from the
baseline up to the apex. From the apex down to the baseline, the slope
becomes decreasingly negative until it returns to zero at the baseline.
If either of the slope values you enter exceeds the slope of the peak
being detected, the software overrides your value and reverts to zero.
Guidelines for
Using These
Parameters
• For typical or symmetrical peaks, use a value of zero.
• For asymmetrical peaks, select values other than zero to better
reflect the beginning and end points.
• A value of zero does not affect the sizing accuracy or precision
of an asymmetrical peak.
Using These
Parameters
Note:
The size of a detected peak is the calculated apex between the
start and end points of a peak and does not change based on your settings.
Baseline
Apex
00
Increasingly
positive slope
(+)
Increasingly
negative slope
(Ð)
To move the… Then… Example
Start point
of a peak
closer to its apex
Change the Slope Threshold for Peak
Start value from zero to a positive
number.
End point of a peak
closer to its apex
Change the Slope Threshold for Peak
End value to a more negative number.

Chapter 3 Algorithms
Slope Threshold Example
GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide 45
Slope Threshold Example
Asymmetrical
Peak
Initial Electropherogram
The initial analysis using a value of 0 for both the Slope Threshold
for Peak Start and the Slope Threshold for Peak End produces an
asymmetrical peak with a noticeable tail on the right side.
Figure 3-12 Electropherogram showing an asymmetrical peak
Adjusting Slope Threshold for Peak End
After reanalyzing with a value of –35.0 for the Slope Threshold for
Peak End, the end point that defines the peak moves closer to its
apex, thereby removing the tailing feature. Note that the only change
to tabular data is the area (peak size and height are unchanged).
.
Figure 3-13 Electropherogram showing the effect of changing
the slope threshold for peak end

Chapter 3 Algorithms
Size-Matching/Size-Calling Algorithm
46 GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide
Size-Matching/Size-Calling Algorithm
Size-Matching
Size-Calling
Algorithm
This algorithm uses a dynamic programming approach that is
efficient (runs in low polynomial time and space) and guarantees an
optimal solution. It first matches a list of peaks from the
electropherogram to a list of fragment sizes from the size standard. It
then derives quality values statistically by examining the similarity
between the theoretical and actual distance between the fragments.
Size-Matching
Algorithm
Example
Figure 3-14 shows an example of how the size-matching/calling
algorithm works using contaminated GeneScan
™
120 size standard
data.
Detected peaks (standard and contamination) are indicated by blue
lower bars along the x-axis. The size standard fragments as
determined by the algorithm (and their corresponding lengths in base
pairs) are designated by the upper green bars. Note that there are
more peaks than size standard locations because the standard was
purposely contaminated to test the algorithm. The algorithm correctly
identifies all the size standard peaks and removes the contamination
peaks (indicated by the black triangles) from consideration. The large
peak is excluded from the candidate list by a filter that identifies the
peak as being atypical with respect to the other peaks.
Figure 3-14 Size-matching example
Bars indicate detected peaks
Bars indicate size standard determined by algorithm

Chapter 3 Algorithms
Size-Calling Methods (Classic and Advanced Modes)
GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide 47
Size-Calling Methods (Classic and Advanced Modes)
Typ es of S iz e
Calling Methods
The GeneMapper Software provides the following size calling methods:
■ Least Squares Method (2nd- and 3rd-Order) . . . . . . . . . . . . . 47
■ Cubic Spline Interpolation Method . . . . . . . . . . . . . . . . . . . . 49
■ Local Southern Method . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 50
■ Global Southern Method . . . . . . . . . . . . . . . . . . . . . . . . . . . . 52
Least Squares Method
Overview Both Least Squares methods (2nd-Order and 3rd-Order) use
regression analysis to build a best-fit size calling curve. This curve
compensates for any fragments that may run anomalously.
Consequently, this method typically results in the least amount of
deviation for all the fragments, including the size standards and the
samples.
Depending on whether you choose the 2nd- or 3rd-Order Least
Squares Method in the Analysis Parameters dialog box, the resulting
size curve is either a quadratic or a cubic function. The software uses
the known standard fragments and the associated data points to
produce a sizing curve based on Multiple Linear Regression.
Advantages Figures 3-15 and 3-16 on page 48 show that in nearly all instances
the mobility of an individual DNA fragment is coincident with the
best curve fit of the entire data set. Stated differently, the mobility of
most DNA fragments is strictly length dependent. This method
automatically compensates for fragments that run anomalously.
The GeneMapper Software calculates a best-fit least squares curve
for all samples, regardless of the size-calling method you choose.
The curve is black in the Standard Sizing Curve window.
Note: The graphs in this section were generated using Version 3.5.1
of the GeneScan
®
Software. The results are similar to those obtained
when you use the GeneMapper Software.

Chapter 3 Algorithms
Size-Calling Methods (Classic and Advanced Modes)
48 GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide
Figure 3-15 2nd-Order Least Squares size calling curve
Figure 3-16 3rd-Order Least Squares size calling curve
Chapter 3 Algorithms
Size-Calling Methods (Classic and Advanced Modes)
GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide 49
Cubic Spline Interpolation Method
Overview
The Cubic Spline method forces the sizing curve through all the
known points of the selected size standard. Although this enforcement
produces exact results for the values of the standards themselves, it
does not compensate for standard fragments that may run anomalously.
Figure 3-17 Cubic Spline Interpolation Method
Possible Local
Sizing Inaccuracy
Mobility of any DNA fragment can be affected by its sequence, and
by secondary and tertiary structure formation. If any internal size
standard fragment has anomalous mobility, the Cubic Spline method
may exhibit local sizing inaccuracy.
For example, assume that a standard fragment is close in molecular
length to an unknown sample fragment. Assume further that the
standard fragment runs anomalously. The Cubic Spline method
assigns the official value to this standard fragment, even though it
may be slightly incorrect. The size of the unknown fragment is then
likely to be calculated incorrectly as well.
Note: This method does not determine the amount of sizing accuracy
error.

Chapter 3 Algorithms
Size-Calling Methods (Classic and Advanced Modes)
50 GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide
Local Southern Method
Overview The Local Southern method determines the sizes of fragments by
using the reciprocal relationship between fragment length and
mobility, as described by E. M. Southern (1979).
Figure 3-18 Local Southern Method
Local Southern
Method Equation
The equation attempts to describe the reciprocal relationship between
the mobility, m, and the length, L0, of the standard fragments.
L = [c/(m–m0)] + L0
How The Local
Southern Method
Works
This method, which is similar to the Cubic Spline method, uses the
four fragments closest in size to the unknown fragment to determine
a best-fit line value. Only the region of the size ladder near the
fragment of unknown length is analyzed.
Note: Size estimates may be inaccurate if any of the standard
fragments run anomalously.

Chapter 3 Algorithms
Size-Calling Methods (Classic and Advanced Modes)
GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide 51
In the Local Southern method:
• The fitting constants of the curve are calculated for each group
of three neighboring points on the standard. A separate curve is
created for each set of three points.
• A curve is then created by using three standard points (two
points below and one point above the fragment), then a fragment
size is determined.
• Another curve is created by looking at an additional set of three
points (one point below and two points above the fragment),
then another value is assigned.
• The two size values are averaged to determine the unknown
fragment length.

Chapter 3 Algorithms
Size-Calling Methods (Classic and Advanced Modes)
52 GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide
Global Southern Method
Overview This method is similar to the Least Squares method in that it
compensates for standard fragments that may run anomalously. The
method creates a best-fit line through all the available points, and
then uses values on that line to calculate the fragment values.
Figure 3-19 Global Southern Method
Global Southern
Method Equations
How the Global
Southern Method
Works
All points in the standard are weighted equally, and the curve is not
constrained to go through any specific point. The software can
analyze a large range of fragment sizes with this method. For best
results, use a standard that brackets all the fragments of interest.
Equation Description
Attempts to describe the reciprocal
relationship between the mobility, m,
and the length, L0, of the standard
fragments.
The fitting constants L0, m0, and c
are calculated by a least-squares fit
to minimize the left side quantity.
Lcmm0–()⁄[]L0+=
iLi c mi m0–()L0+()⁄[]–{}
2
∑

Chapter 3 Algorithms
Allele-Calling Algorithms
GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide 53
Allele-Calling Algorithms
Overview Final allele calls are based on a consensus between a variety of
different allele-calling algorithms. Each caller has a different design
philosophy such that it excels in a particular data regime but not in
others. Allele-calling algorithms involve envelope detection,
optimization of parametric models, and rule-based systems.
Types of Allele-
Calling Methods
The GeneMapper Software provides following allele-calling methods:
■ Microsatellite Analysis Methods . . . . . . . . . . . . . . . . . . . . . . 54
■ SNPlex™ System Analysis Methods . . . . . . . . . . . . . . . . . . . 55
– Rules Genotyping Algorithm (see page 55)
– Model Genotyping Algorithm (see page 56)

Chapter 3 Algorithms
Allele-Calling Algorithms
54 GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide
Microsatellite Analysis Methods
Example Output of
Different Allele-
Calling Algorithms
Figure 3-20 on page 54 shows an example of three different allele-
calling algorithms for 16 samples. User annotations are indicated by the
(red) circles, and allele caller outputs are indicated by the (green, black,
and blue) asterisks. Note that consensus between multiple callers
virtually ensures that the calls are correct. In samples (i) and (p), the
algorithms have not made a call because they determined that the data
are too complex to act on. Here the blue asterisks show the calls
transmitted to the user. Low-quality values are reported because in both
cases the first algorithm did not call, and in (i), the black caller does not
agree with the blue. Despite these conditions, however, the calls are
correct. The low-quality values alert the user to potential problems such
as the spurious peak in (i) and the high background in (p).
Figure 3-20 The effect of three different allele-calling algorithms
on 16 different samples

Chapter 3 Algorithms
Allele-Calling Algorithms
GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide 55
SNPlex
™
System Analysis Methods
Overview The GeneMapper Software provides the following allele-calling
methods for SNPlex System Analysis:
■ Rules Genotyping Algorithm . . . . . . . . . . . . . . . . . . (see below)
■ Model Genotyping Algorithm . . . . . . . . . . . . . . . . . . . . . . . . 56
Rules Genotyping
Algorithm
The Rules (Maximum-Likelihood or ML) SNP genotyping algorithm
consists of several processes: genotype cluster identification, sample
cluster classification, and confidence value assignment.
Figure 3-21 Rules algorithm block diagram
Because of small systematic differences in assay performance
between the alleles in a SNP, fixed classification boundaries between
the clusters do not give accurate results for most, if not all, current
SNP detection platforms. Clustering, like all calibration methods, can
correct for systematic errors, but not random errors. Random errors
are estimated and assigned appropriate p-values.
Input: Allele 1 and 2 Intensities
Allele Intensity Correction
Estimate Model Parameters
• Number of clusters
• Mean, variance of each cluster
• Allele frequency
Output: Genotypes and
Quality Values for all Points
and Plate Score (optional)
Filter Samples and Recluster
Done
Compute Most Likely Genotype
and Confidence Values
• Outlier probability
Detect Outliers
Resampling
(Exclude Outliers)
First Pass
Second Pass
Second Pass

Chapter 3 Algorithms
Allele-Calling Algorithms
56 GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide
Genotype Cluster Identification
The cluster identification stage uses
a priori
knowledge of the
intensity relationship and frequency distribution of SNP alleles to
construct a most likely model of the SNP assays generating in a given
data set. Systematic parameters such as allele assay strength and
other chemistry factors are estimated. If the most likely model fit is
too poor, the entire data set is rejected.
Sample Cluster Classification and Confidence Value Assignment
The best fit model is used to classify each point into its most likely
class, or genotype. The likelihood of class membership (genotype)
can be derived in the same step.
Post Processing, Filtering, and Reiterations
Genotype and sample filters, bootstrap resampling, and several
reiterations are used to ensure the accuracy of the model fit and
classifications.
Model Genotyping
Algorithm
The Model genotyping algorithm consists of several processes:
normalization, allele calling, and confidence value generation.
Normalization
Normalization is a run-based algorithm that removes systematic
variation of peak height by allele and sample. This process is
accomplished by comparing the data to a model in which the sum of
allele peaks for any given sample is a constant. Over many repeated
measurements of a given sample or allele, systematic variations from
this a priori model can be quantified and later removed. In the
process of normalization, an overall scale factor can be selected,
which can be set to one. This utility is demonstrated in Figure 3-22
on page 57 where the output from 20 runs can be overlaid to
understand the random, as opposed to systematic, behavior of
signals. For each run 2 ∞ 48 ∞ 96 = 9216 measurements were
normalized with 193 parameters, or approximately one parameter for
each 48 measurements.

Chapter 3 Algorithms
Allele-Calling Algorithms
GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide 57
Figure 3-22 Pre- and post- normalization data. Shown are 80,000
measurements of SNPlex System data overlaid in normalized allele
coordinates. In the left plot, data are normalized only by run. In the
right, the data are fully normalized by sample and allele.
Genotyping
The random error remaining in the data after normalization is relatively
small when compared with the separation of the genotype coordinates,
as seen in Figure 3-23. Each measurement is assigned the genotype for
which it has the highest probability.
Figure 3-23 Training data distribution
Allele 2
Homozygotes
Heterozygotes
Allele 1
Homozygotes

Chapter 3 Algorithms
Allele-Calling Algorithms
58 GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide
Final Genotyping and Confidence Value Assignment
The confidence value is a quantitative estimate of the probability of a
correct call (that is one minus the estimated error rate). This single
parameter can be used to optimize the trade-off between throughput
and accuracy for a particular data set, as shown in Figure 3-24.
Figure 3-24 Accuracy-throughput trade off
100
90
80
70
60
50
40
30
20
10
100
99.8
99.6
99.4
99.2
99.0
0.65 0.70 0.75 0.80 0.85 0.90 0.95 1.0
Quality Value
Fraction Accuracy (%)Net Accuracy (%)
Fail Pass

GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide 59
Glossary
ABB (Automatic
Bin Builder)
The first step in accurate allele assignment. After sample files are
collected, bins are created by the ABB based on the chosen panel
information and successive allele calls from sample file collection.
As each sample file in the collection is processed, the bin definitions
are refined to reflect the actual data.
access control list (See Security Group)
Admin profile A pre-configured profile that cannot be removed and that has execute
access to all functions. Initially associated with the “admin” user. (A
user must always have an assigned profile.)
Admin security
group
A pre-configured security group that cannot be removed. This
security group has been granted all rights to all data, to provide a way
for at least one user to have “admin” access to all data.
Admin User Group A pre-configured user group that cannot be removed and that is
always associated with the Admin security group.
Administrator User A pre-configured user that cannot be removed and that is always
associated with the Admin user group.
AFLP
®
Amplified Fragment Length Polymorphism. A DNA fingerprinting
technique that allows the comparison of the DNA from different
organisms. DNA fragments of varying lengths are created by
cleaving an organism’s DNA with restriction enzymes; a specific
subset of these fragments are amplified and analyzed for comparison
purposes.

Glossary
60 GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide
Algorithm A set of ordered steps for solving a problem, such as a mathematical
formula or the instructions in a program. The terms algorithm and
logic are synonymous, where both refer to a sequence of steps to
solve a problem. However, an algorithm is an expression that solves a
complex problem rather than the overall input-process-output logic
of typical business programs.
All User group A user group that contains all users. A user cannot be disassociated
from the user group.
allele One of two or more alternate forms of a marker or gene.
Reference alleles are all alleles or bins created in a bin set in the
GeneMapper Software. They are denoted by a red cross hatch in
the Panel Manager.
Project alleles are all alleles detected in sample data in a project in
the GeneMapper Software. They are denoted by a blue asterisk in the
Panel Manager.
allele call Identification of the specific allelic form of a marker.
allele-calling Identification of alleles based on bin definitions; genotyping;
GeneMapper
®
Software analysis
allelic ladder A sample of DNA containing most possible alleles for a specific
marker or set of markers. Used to create a sample file that the
GeneMapper Software can use to genotype or make allele calls on
sample data.
Within the GeneMapper Software, you select a Sample Type of
Allelic Ladder for the sample file generated using an allelic ladder.
analysis method A collection of user-defined parameters that determine the bin set and
analysis algorithms.
analysis
parameters
A collection of user-defined settings (including analysis method, size
standard, and panel) that determine the sizing and genotyping
algorithms used by the GeneMapper Software to analyze all sample
files in a project. Also called project settings.

Glossary
GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide 61
association Two identifiers combined are said to be associated. A user can be
associated with a user group. A user group associated with a security
group yields a set of data rights.
audit event A single permanent change to one or more attributes of an object.
Includes creating a new instance of an object or deleting an exiting
one.
audit map An object associated with an object type; used to tell the audit
component how to audit an object type.
audit object A collection of data defined by an application. Also referred to as an
object.
audit record The description of a single audit event.
autopanelizer A feature that uses reference data generated by the Primer Focus
®
kit
to quickly define new SNP markers and bin sets.
bin Within the GeneMapper Software, a fragment size (bp) or basepair
range and dye color that define an allele within a marker. You create a
bin for each possible allele associated with a marker.
bin set Within the GeneMapper Software, a collection of bins (allele
definitions), typically specific to a set of experimental conditions,
usually an instrument. Bin sets are available inside a kit.
cache An “in memory” representation of the access control data. The
Admin Tool modifies the data in the cache. When the Admin Tool or
Admin API issues the “save” command, the data in the cache are
written to the data store.
challenge A term from user authentication indicating that the user is asked to
provide identification (typically by entering a password).
chromosome A long stretch of coiled strands of DNA and proteins containing
many genes. Human DNA is contained within 23 pairs of
chromosomes.

Glossary
62 GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide
control See “positive control” on page 64.
See “negative control” on page 64.
Control Security
Group
The security group assigned to an Access Control administrative
identifier (user, user group, security group, profile). This security
group is used to determine access by a user to the administrative data
in the Administrative GUI and API.
data access control The part of access control that administers access to user data.
Data group (See Security Group)
data rights Properties that define the type of access a user has to a piece of data.
database One form of offline storage.
diploid Having two sets of chromosomes and, therefore, having two alleles
per marker or gene. Human cells (other than egg and sperm cells) are
diploid.
dye set A set of four to five different colored dyes. A specific dye set is used
to label DNA fragments or markers in matrix standards, installation
standards, and chemistry kits.
electropherogram A graphical representation of the intensity (y-axis) of bands produced
in a single gel lane or capillary as a function of time (x-axis).
electrophoresis A technique used to separate molecules by using an electric field to
pass those molecules through a porous matrix.
gene The basic unit of heredity that carries the genetic information for a
given RNA molecule or protein.
genome All the DNA contained in an organism or cell, including both the
chromosomes in the nucleus and the DNA in the mitochondria.
genotype The set of allele calls for specific markers or genes within an
organism. (noun)
To determine the allele calls for specific markers or genes within an
organism. (verb)

Glossary
GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide 63
haploid Having one set of chromosomes and, therefore, having one allele per
marker or gene. Human egg and sperm cells are haploid.
heterozygous Having two different alleles for a specific marker or gene.
homozygous Having two identical alleles for a specific marker or gene.
installation
standard
A collection of known genetic markers, labeled with dyes from a
specific dye set, used to test the function of a genetic analyzer.
kit Within the GeneMapper Software, a group of panels.
LMS Linkage Mapping Set; Applied Biosystems chemistry using
dinucleotide repeat microsatellite markers
locus The chromosomal location of a genetic marker or gene.
marker A known segment of DNA that has two or more allelic forms. A
marker exists at a known chromosomal loci and can be a gene or a
non-gene. See also microsatellite and SNP.
Within the GeneMapper Software:
A microsatellite marker is defined by a name, fragment size range
(bp), dye color, and repeat length.
A SNaPshot kit analysis marker is defined by a name and fragment
size range (bp).
matrix standard A collection of known DNA fragments, labeled with four to five
different colored dyes from a specific dye set. The matrix standard is
run on a genetic analyzer and used for spectral calibration of sample
fragments that are run on the same instrument and labeled with the
same dye set.
microsatellite Microsatellite markers, also known as short tandem repeats (STRs),
are polymorphic DNA loci consisting of a repeated nucleotide
sequence. The repeat sequence can be from 2 to 7 base pairs long.
The number of repeat units varies within a population, thereby
creating multiple alleles for a microsatellite locus.

Glossary
64 GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide
negative control A blank sample that contains no DNA, but all other reagents used in
the experiment. It can indicate if any contamination came from
sample preparation.
In the Sample tab of GeneMapper Software, you select a Sample
Type of Negative Control for the sample file generated from the
negative control. Additionally, in the Panel Manager you define the
negative control when creating markers.
panel A group of markers. Within the GeneMapper Software, you associate
a panel with a bin set to provide bin definitions for the markers.
phenotype The physical manifestation of a genotype.
polymorphism Differences between organisms’ or individuals’ DNA. Variations of
allele calls.
positive control A sample that contains DNA with known alleles for specific markers.
Its purpose is to verify that the PCR amplification, electrophoresis,
and GeneMapper Software analysis worked correctly.
In the Sample tab of GeneMapper Software, you select a Sample
Type of Positive Control for the sample file generated from the
positive control. Additionally, in the Panel Manager you define the
positive control when creating markers.
primer A single-stranded piece of DNA or RNA that anneals to a
complementary section of DNA and serves as a starting point for
chain extension by DNA polymerase.
primer focus Within the GeneMapper Software, you select a Sample Type of
Primer Focus for the sample file generated using the Primer Focus
kit. The GeneMapper Software uses this file to automatically create
bins using the Auto Panel feature.
Primer Focus
®
kit An Applied Biosystems kit containing reagents used to create and
amplify all four possible alleles of any SNP marker. The kit allows
you to take advantage of the Auto Panel feature in the GeneMapper
Software to automatically create bins for each allele in a SNaPshot
kit analysis.

Glossary
GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide 65
probe A DNA or RNA fragment that has been labeled in some way (for
example, fluorescent or radioactive), then used in a molecular
hybridization assay to identify DNA or RNA sequences that are the
same or closely related to it in sequence.
profile An identifier that gives an administrator the ability to grant or revoke
access to functions.
project Within the GeneMapper Software, a collection of sample files and
the analysis parameters for genotyping them.
project settings See “analysis parameters” on page 60.
project settings Parameters set by the user to prepare a project for analysis.
reference samples All or a subset of the actual samples. The reference samples typically
contain all of the alleles present in the sample set and are used to
create a bin for each allele within the GeneMapper Software.
rights Properties that define whether a user has access to data or a function.
Security group An identifier that can be associated with a user group to confer a set
of data rights.
Security ID The universal identifier of the security group and the preferred name
of the column in an application table that holds the security group ID.
SFNF Sample File Not Found
silent auditing Automatic audit record creation (without prompting of the user).
size match editor A window in GeneMapper
®
Software that allows users to examine
size-standard electropherograms, edit the identification of size-
standard peaks, and view the size-calling curve.
size standard A collection of DNA fragments of known lengths within a range (for
example, 50 to 400 bp) all tagged with the same dye. The size
standard is co-injected into the genetic analyzer capillary with the
sample, then used to size the sample data. All Applied Biosystems
size standards are labeled with a red or orange dye.

Glossary
66 GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide
SNaPshot
®
kit An Applied Biosystems kit containing reagents used to PCR amplify
any SNP markers, using single-base-extension technology. Sample
files can then be sized and genotyped by using a SNaPshot analysis
in the GeneMapper Software.
SNaPshot
®
System
Multiplex Analysis
Primer extension-based chemistry for SNP validation
SNP Single Nucleotide Polymorphism. A marker consisting of a single
base pair that varies, thereby creating up to four alleles of the marker.
In this document, SNP refers to SNaPshot
®
system markers and
SNPlex
™
systems
SNP Single-Nucleotide Polymorphism (used)
SNPlex™ System High-throughput assay for genotyping.

GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide 67
Index
A
ABB (automatic bin builder), definition 59
access control list, definition 59
admin profile, definition 59
admin security group, definition 59
admin user group, definition 59
admin user, definition 59
ADO (Allele Display Overflow) PQV 8
AE (Allele Edit) PQV 8
algorithms
allele-calling 36, 53
binning 36
definition 60
overview 36
peak detection 36
size-matching 36
all user group, definition 60
Allele Display Overflow PQV 8
Allele Edit PQV 8
Allele Number PQV 9
allele-calling
algorithm 53
definition 60
AN (Allele Number) PQV 9
analysis method, definition 60
Applied Biosystems
contacting viii
customer feedback on documentation viii
Information Development
department viii
Technical Support viii
association, definition 61
assumptions for using this guide v
audit event, definition 61
audit map definition 61
audit object, definition 61
audit record, definition 61
autopanelizer, definition 61
B
BD (Broad Peak) PQV 9
BIN (Out of Bin Allele) PQV 10
bold text, when to use v
Broad Peak PQV 9
C
cache, definition 61
CC (Control Concordance) PQV 11
challenge, definition 61
Control Concordance PQV 11
control security group, definition 62
conventions
bold text v
for describing menu commands v
IMPORTANTS! vi
in this guide v
italic text v
Notes vi
user attention words vi
Cross Talk PQV 32
customer feedback, on Applied Biosystems
documents viii
D
data access control, definition 62
data flow, genotyping algorithms 36
data group, definition 62
Index
68 GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide
data rights, definition 62
database, definition 62
documentation, related vii
Double Peak PQV 12
DP 12
DP (Double Peak) PQV 12
E
EPT data, displaying 5
examining peak definitions 39
example output of different allele-calling
algorithms 54
G
Genotype Quality PQV 13
genotyping algorithms 36
GQ (Genotype Quality) PQV 13
H
help, online, accessing vii
I
Information Development department,
contacting viii
italic text, when to use v
L
LMS, definition 63
Low Peak Height PQV 15
LPH (Low Peak Height) PQV 15
M
Marker Quality 14
Matrix Not Found PQV 16
menu commands, conventions for
describing v
MNF (Matrix Not Found) PQV 16
MSDSs, obtaining viii
N
Narrow Bin PQV 18
NB (Narrow Bin) PQV 18
numeric quality metrics, displaying 7
O
OBA (One Basepair Allele) PQV 18
Offscale PQV 19
One Basepair Allele PQV 18
online help, accessing vii
optimizing peak detection sensitivity 41
OS (Offscale) PQV 19
Out of Bin Allele PQV 10
Overlap PQV 20
OVL (Overlap) PQV 20
P
peak definitions, examining 39
peak detection 38
effects of extreme settings 43
guidelines for use 39
optimizing sensitivity 41
parameters 38
peak window size 38
polynomial degree 38
slope threshold 44
peak detection sensitivity, reducing window
size 41
Peak Height Ratio PQV 21
PHR (Peak Height Ratio) PQV 21
polynomial degree 38
peak detection 38
varying 39
window size value 40
possible local sizing inaccuracy 49
Process Quality Values (PQV) 2 to 32
displaying as numbers 7
profile, definition 65
Index
GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide 69
project settings, definition 65
R
Raw Data, displaying 5
reducing window size and increasing
polynomial degree 42
rights, definition 65
S
Sample File Not Found PQV 22
sample information, displaying 5
security group, definition 65
security ID, definition 65
SFNF (Sample File Not Found) PQV 22
Sharp Peak PQV 23
SHP (Sharp Peak) PQV 23
silent auditing, definition 65
Single Peak Artifact PQV 24
size calling 53
advanced method 47
classic method 47
cubic spline interpolation method 49
global southern method 52
least square method 47
local southern method 50
overview 47
size match editor, definition 65
Size Standard Not Found PQV 23
size-matching/size-calling algorithm 46
Sizing Quality PQV 25
slope threshold
asymmetrical peak 45
peak end parameters 44
peak start parameters 44
SNaPshot
®
analysis, definition 66
SNF (Size Standard Not Found) PQV 23
SNP
definition 66
SNPlex™ system analysis, definition 66
SP (Split Peak) PQV 24
SPA (Single Peak Artifact) PQV 24
Spectral Pull-Up PQV 25
Split Peak PQV 24
SPU (Spectral Pull-Up) PQV 25
SQ (Sizing Quality) PQV 25
T
Technical Support, contacting viii
text conventions v
training, information on viii
U
user attention words, described vi
V
varying polynomial degree 39
W
window size value, increasing 40
X
XTLK (Cross Talk) PQV 32
Index
70 GeneMapper
®
Software Version 4.1 Reference and Troubleshooting Guide

Part Number 4403673 Rev. A 04/2009
Technical Resources and Support
For the latest technical resources and
support information for all locations,
please refer to our Web site at
www.appliedbiosystems.com/support
Applied Biosystems
850 Lincoln Centre Drive
Foster City, CA 94404 USA
Phone 650.638.5800 | Toll Free 800.345.5224
www.appliedbiosystems.com
