
GE47CH05-Sabeti ARI 29 October 2013 12:12
Detecting Natural Selection
in Genomic Data
Joseph J. Vitti,
1,2
Sharon R. Grossman,
2,3,4
and Pardis C. Sabeti
1,2
1
Department of Organismic and Evolutionary Biology, Harvard University, Cambridge,
2
Broad Institute of MIT and Harvard, Cambridge, Massachusetts 02142
3
Department of Systems Biology, Harvard Medical School, Boston, Massachusetts 02115
4
Department of Biology, Massachusetts Institute of Technology, Cambridge,
Massachusetts 02139
Annu. Rev. Genet. 2013. 47:97–120
The Annual Review of Genetics is online at
genet.annualreviews.org
This article’s doi:
10.1146/annurev-genet-111212-133526
Copyright
c
2013 by Annual Reviews.
All rights reserved
Keywords
population genetics, adaptation, selective sweeps, genome scans,
evolutionary genomics
Abstract
The past fifty years have seen the development and application of nu-
merous statistical methods to identify genomic regions t hat appear to
be shaped by natural selection. These methods have been used to in-
vestigate the macro- and microevolution of a broad range of organisms,
including humans. Here, we provide a comprehensive outline of these
methods, explaining their conceptual motivations and statistical inter-
pretations. We highlight areas of recent and future development in
evolutionary genomics methods and discuss ongoing challenges for re-
searchers employing such tests. In particular, we emphasize the impor-
tance of functional follow-up studies to characterize putative selected
alleles and the use of selection scans as hypothesis-generating tools for
investigating evolutionary histories.
97
Annu. Rev. Genet. 2013.47:97-120. Downloaded from www.annualreviews.org
by Harvard University on 11/26/13. For personal use only.
Click here for quick links to
Annual Reviews content online,
including:
• Other articles in this volume
• Top cited articles
• Top downloaded articles
• Our comprehensive search
Further
ANNUAL
REVIEWS
GE47CH05-Sabeti ARI 29 October 2013 12:12
INTRODUCTION
As humans and other organisms moved to
inhabit every part of the world, they were ex-
posed to myriad new environments, diets, and
pathogens, and forced to adapt, leading to the
great diversity we observe today. Uncovering
the mechanism of this diversification has for
years fascinated scientists and nonscientists
alike. In 1858, Darwin and Wallace gave
grounds for species evolution when they
articulated the principle of natural selection,
the idea that beneficial traits—those that
improve an individual’s chances to survive and
reproduce—tend to become more frequent in
populations over time.
Scientists have continued to search for
evidence of evolution and for the specific
adaptations that underlie it. Animal and plant
breeders were some of the first to identify
traits that are evolving, as they witnessed
dramatic changes in their stock through
artificial selection. Haldane uncovered the first
adaptive trait in humans when he observed
that many diseases of red blood cells seemed
to be distributed in regions where malaria was
endemic (48). Haldane’s malaria hypothesis
was confirmed by Allison a few years later,
when he demonstrated that the sickle cell
mutation in the Hemoglobin-B gene (HBB) was
the target of selection for malaria resistance (4).
The ability to assess evidence for selection
at the genetic level represented a breakthrough
for this pursuit. Computational analysis of pop-
ulation genetic data sets provides a statistically
rigorous way to infer the action of natural selec-
tion; in this way, the field of evolutionary genet-
ics represents an antidote to the preponderance
of speculative just-so stories that some biolo-
gists have lamented (42). Moreover, it demon-
strates the full realization of the modern syn-
thesis: Darwinian concepts of selection have
been rendered quantitative and measurable in
real populations, thanks to methodological and
technological advances (1).
Through evolutionary genetics, many adap-
tive traits have been elucidated, from lactase
persistence and skin pigmentation in humans
(90, 125) to coat color in field mice (81) to
armored plates in stickleback fish (64). These
instances were all identified using a forward
genetics approach, in which a phenotype was
first hypothesized to be adaptive and the un-
derlying loci were then identified. With on-
going advancements in genomic technology,
we can now go further, from testing evidence
for selection on putative adaptive traits to un-
covering candidate genetic regions through
genome scans. This transition from hypothesis-
testing to hypothesis-generating science has
been made possible both by the new data (e.g.,
genome sequences from increasing numbers of
species and genome-wide variation data) and by
increasingly sophisticated tools that allow us to
make sense of this deluge of data and to fine-
map evidence of selection to individual candi-
date variants.
Identifying such candidates is significant not
only because they demonstrate evolution and
shed light on species histories but also because
they represent biologically meaningful varia-
tion. Given that selection operates at the level of
the phenotype, alleles showing evidence of se-
lection are likely to be of functional relevance.
Thus, alleles implicated in selection studies are
often linked either to resistance to infectious
diseases, as pathogens are believed to represent
one of the strongest selective pressures acting
on humans (40), or to noninfectious genetic
diseases, such as those associated with autoim-
mune diseases or metabolic disorders (54).
Further breakthroughs in genomic anno-
tation, genome manipulation technology, and
high-throughput molecular biology are be-
ginning to allow researchers to progress from
candidate variants to functionally elucidated
instances of evolution. Taken together, all of
these advancements present a path to realizing
the full potential of evolutionary genomics
in shedding light on species histories and
uncovering biologically meaningful variation.
Modes of Selection
Natural selection is based on the simple obser-
vation that fitness-enhancing traits, i.e., those
98 Vitti
·
Grossman
·
Sabeti
Annu. Rev. Genet. 2013.47:97-120. Downloaded from www.annualreviews.org
by Harvard University on 11/26/13. For personal use only.

GE47CH05-Sabeti ARI 29 October 2013 12:12
Heterozygote
advantage: atrendin
which the fitness of a
heterozygote is greater
than that of either
homozygote. Also
referred to as
overdominance
Frequency-
dependent selection:
a trend in which the
fitness of a given
genotype is correlated
with its prevalence in
the population (e.g., if
an allele is
advantageous when it
is rare)
Codominance:
condition in which
multiple alleles are
dominant; the
heterozygote expresses
phenotypes associated
with both alleles
that improve an organism’s chance of survival
or reproductive success in its environment, are
more likely to be passed on to that organism’s
offspring and therefore increase in prevalence
in the population over time. I n the genomic
era, selection refers to any nonrandom, dif-
ferential propagation of an allele as a conse-
quence of its phenotypic effect. There are many
specific modes of selection that have been de-
scribed, some of which share conceptual over-
lap, and some of which are referred to by multi-
ple names. In this section, we briefly define the
different modes of selection that we employ in
our discussion (85).
Most simply, selection may act in a direc-
tional manner, in which an allele is favored and
so propagated (positive selection) or disfavored
(negative selection, also called purifying selec-
tion). Random mutations are more likely to be
deleterious than beneficial, so many novel al-
leles are immediately subject to negative selec-
tion and become removed from the gene pool
before they can achieve detectable frequency
within the population. This ongoing removal
of deleterious mutations is a form of negative
selection referred to as background selection.
In genetic regions under strong background se-
lection, mutations are quickly removed from
the gene pool, resulting in highly conserved
stretches of the genome (i.e., regions where
variation is not observed).
More subtle configurations of positive and
negative selection give rise to other common
evolutionary trends, particularly (although not
exclusively) in diploid and polyploid organisms,
where the phenotype depends on the interac-
tion of multiple alleles at the same locus. One
such phenomenon is balancing selection, in
which multiple alleles are maintained at an ap-
preciable frequency within the gene pool. This
may happen as the result of, for example, het-
erozygote advantage (i.e., overdominance) or
frequency-dependent selection (20). If the alle-
les being maintained conduce to opposing phe-
notypic effects—for example, if large and small
body sizes are maintained within the population
to the exclusion of intermediate sizes—then the
trend is often further described as diversifying
or disruptive selection. By contrast, when inter-
mediate phenotypic values are favored, whether
by balancing selection of codominant alleles or
by positive selection of alleles that underlie in-
termediate phenotypes, the trend is called sta-
bilizing selection.
This diversity of modes of selection notwith-
standing, much research in recent years has fo-
cused on the development of genomic methods
to identify positive selection. One reason for
this emphasis on positive selection is practical:
Whereas negative selection is primarily observ-
able in highly conserved regions and balancing
selection’s effect on the genome is often subtle,
positive selection leaves a more conspicuous
footprint on the genome that can be detected
using a number of different approaches. An-
other reason for the interest in positive selection
is theoretical: Positive selection is understood
to be the primary mechanism of adaptation
(i.e., the genesis of phenotypes that are apt for
a specific environment or niche), which in turn
poses great theoretical interest to researchers
(1).
Here, we discuss the various approaches that
have been used to identify positive selection
while also indicating the ways that these meth-
ods may be used to detect and classify instances
of other modes of selection (Table 1). These
approaches typically use summary statistics
to compare observed data with expectations
under the null hypothesis of selective neutrality
(see sidebar, Selection and Neutrality).
We begin by discussing methods based
on comparisons of different species and their
relative rates of genetic change. These methods
are most often used to identify selective events
that took place within the deep past and that
reflect macroevolutionary trends that occur as a
result of selection between, rather than within,
species. We then turn our attention to pop-
ulation genetics methods used to identify mi-
croevolutionary selective events within species.
Variants identified by these latter methods are
believed to underlie local adaptations in hu-
mans following the out-of-Africa migration and
www.annualreviews.org
•
Detecting Natural Selection in Genomic Data 99
Annu. Rev. Genet. 2013.47:97-120. Downloaded from www.annualreviews.org
by Harvard University on 11/26/13. For personal use only.

GE47CH05-Sabeti ARI 29 October 2013 12:12
Table 1 An overview of common approaches for detecting selection
Approach Intuition Representative tests References
Methods for
macroevolution
K
a
/K
s
(alsoreferredtoas
d
N
/d
S
or ω)
(43, 60)
Gene-based methods Synonymous substitutions are
(assumed to be) selectively neutral.
Thus, they tell us about the
background rate of evolution. If the
rate of nonsynonymous substitution
differs significantly, it is suggestive
of selection.
McDonald-Kreitman test
(MKT)
(27, 78)
Other rate-based
methods
Levels of polymorphism and
divergence should be correlated
(because both are primarily
functions of the mutation rate)
unless selection causes one to
exceed the other.
Hudson-Kreitman-
Aguad
´
e (HKA) test
MKT
(59, 135)
Regions that undergo accelerated
change in one lineage but are
conserved in related lineages are
probable candidates for selection.
Identification of
accelerated regions
(14, 77, 100,
102, 116)
Methods for
microevolution
Ewens-Watterson test (30, 133)
Tajima’s D and derivatives (38, 39, 122,
123)
Frequency-based
methods
In a selective sweep, a genetic variant
reaches high prevalence together
with nearby linked variants (high-
frequency derived alleles). From
this homogenous background, new
alleles arise but are initially at low
frequency (surplus of rare alleles).
Fay & Wu’s H (33)
Long-range haplotype
(LRH) test
(111, 141)
Long-range haplotype
similarity test
(52)
Integrated haplotype
score (iHS)
(131)
Cross-population
extended haplotype
homozygosity
(XP-EHH)
(113)
Linkage disequilibrium
decay (LDD)
(132)
Linkage
disequilibrium–
based
methods
Selective sweeps bring a genetic
region to high prevalence in a
population, including the causal
variant and its neighbors. The
associations between these alleles
define a haplotype, which persists in
the population until recombination
breaks these associations down.
Identity-by-descent (IBD)
analyses
(15, 50)
Lewontin-Krakauer test
(LKT)
(11, 31, 73, 129)
Locus-specific branch
length (LSBL)
(117)
Population
differentiation–
based
methods
Selection acting on an allele in one
population but not in another
creates a marked difference in the
frequency of that allele between the
two populations. This effect of
differentiation stands out against
the differentiation between
populations with respect to neutral
(i.e., nonselected) alleles.
hapFLK (32)
(Continued )
100 Vitti
·
Grossman
·
Sabeti
Annu. Rev. Genet. 2013.47:97-120. Downloaded from www.annualreviews.org
by Harvard University on 11/26/13. For personal use only.

GE47CH05-Sabeti ARI 29 October 2013 12:12
Table 1 (Continued)
Approach Intuition Representative tests References
Composite methods Composite likelihood
ratio (CLR)
(67, 68, 87, 89)
Combining test scores for multiple
sites across a contiguous region
can reduce the rate of false
positives.
Cross-population
composite likelihood
ratio (XP-CLR)
(22)
Combining multiple independent
tests at one site can improve
resolution and distinguish causal
variants. Different tests can
provide complementary
information.
DH test (138, 139)
Composite of multiple
signals (CMS)
(44, 45)
Homologs: traits or
sequences that are
similar in disparate
groups because of
common ancestry
Synonymous: a
change in the protein-
coding region of a
gene that does not
change the amino acid
encoded
thus have become the subject of much research
toward understanding human evolution and
history (112).
DETECTING SELECTION AT
THE MACROEVOLUTIONARY
LEVEL
Methods to detect selection at the macroevolu-
tionary level typically hinge on comparisons of
homologous traits or sequences among related
taxa (Figure 1a). These methods identify se-
quences that are likely to be functional (either
because they code for proteins or because they
are conserved among different species) and
then search for lineage-specific accelerations
in the rate of evolution. Such accelerations are
indicated by an excess of substitutions relative
to the baseline mutation rate, which can be
calculated either from the rate of synonymous
mutations (which are generally considered
neutral, but see Reference 21) or from the
overall rate of substitutions between species.
Gene-Based Methods
Perhaps the best-known statistic for detecting
selection is K
a
/K
s
,alsoreferredtoasd
N
/d
S
or
ω (Figure 1b). This statistic compares the rate
of nonsynonymous substitutions per site (i.e.,
per potential nonsynonymous change) with the
rate of synonymous substitutions per site (i.e.,
per potential synonymous change) (60). Be-
cause synonymous changes are assumed to be
functionally neutral (silent), their substitution
rate provides a baseline against which the rate
of amino acid alterations can be interpreted. A
relative excess of nonsynonymous substitutions
indicates ongoing (or recently ended) positive
selection favoring novel protein structures (or
else a cessation of negative selection against
protein alterations; see section, Challenges
in Applying Statistical Tests for Selection).
This is summarized by a value of K
a
/K
s
greater than 1, whereas smaller values indicate
SELECTION AND NEUTRALITY
Kimura’s neutral theory of molecular evolution held that the vast
majority of genetic change is attributable to genetic drift rather
than Darwinian selection (69). However, as researchers began to
develop methods to distinguish neutral from adaptive change in
the genome, many came to reject the stronger versions of the
neutral theory and turned their attention toward quantifying the
relative contributions of drift and selection to molecular evolution
(71, 120).
Importantly, however, the neutral theory enabled the devel-
opment of tests for selection by assisting in the sophistication
of models of genetic drift. In many tests for selection (neutrality
tests), r esearchers compare empirical data against data generated
by simulations of drift, which serve as a null hypothesis. Other
neutrality tests may use background rates of change inferred from
whole-genome analyses to furnish a null hypothesis.
In this review, we focus our discussion on the wide range of
tests for selection that have been developed and their applica-
tions. Readers interested in the selectionist-neutralist debate are
encouraged to consult recent reviews on the subject (7, 33, 83).
www.annualreviews.org
•
Detecting Natural Selection in Genomic Data 101
Annu. Rev. Genet. 2013.47:97-120. Downloaded from www.annualreviews.org
by Harvard University on 11/26/13. For personal use only.

GE47CH05-Sabeti ARI 29 October 2013 12:12
a
TGCAGAGTAAGACCT
TGCAGAGTAAGACCT
TGCAGAGTAAGACCT
TGTAGCG CCAGACAT
Species 1
Species 2
Species 3
Species 4
Ancestral sequence
Species 4
Nonsynonymous substitutions
Synonymous substitutions
K
a
K
s
> 1?
b
Time
Genomic position
Positive selection
Divergence >
polymorphism
Substitution Mutation
c
Neutrality
Divergence
polymorphism
TCCAGAATGAGACGT
Figure 1
Methods for detecting selection at the macroevolutionary level. (a) Traits that are conserved across many
clades of a phylogeny but that show extreme differentiation in one or a few lineages are likely candidates for
selection. (b) Metrics such as K
a
/K
s
compare the rate of nonsynonymous (i.e., amino acid–altering)
substitutions in a lineage to the rate of synonymous substitutions, which are assumed to be selectively
neutral. (c) The McDonald-Kreitman test and the Hudson-Kreitman-Aguad
´
e test hinge on the intuition that
levels of interspecies divergence and of intraspecies polymorphism are governed by the mutation rate and are
correlated unless selection or some other force (e.g., fluctuations in population size) is at play.
Nonsynonymous: a
change in the protein-
coding region of a
gene that alters the
amino acid encoded
ongoing negative selection against deleterious
mutations and the consequent preservation
of protein structure. These methods may
also be applied across an entire open reading
frame or some subdivision thereof (down to
an individual codon), as different regions of a
protein may be subject to different selective
pressures (136). Various models for calculating
synonymous and nonsynonymous substi-
tution rates take into account the different
probabilities of different mutations (e.g.,
transitions are more likely than transversions)
102 Vitti
·
Grossman
·
Sabeti
Annu. Rev. Genet. 2013.47:97-120. Downloaded from www.annualreviews.org
by Harvard University on 11/26/13. For personal use only.

GE47CH05-Sabeti ARI 29 October 2013 12:12
Codon usage bias:
the tendency of an
organism’s genome to
more commonly have
a certain codon for a
given amino acid than
any of its synonymous
counterparts
as well as the possibility of unobserved changes
(e.g., if one species undergoes two sequential
mutations at the same site) and codon usage
bias (43).
The McDonald-Kreitman test (MKT)
builds upon this method by utilizing not
only interspecies divergence data but also in-
traspecies diversity data (78). Essentially, the
MKT compares two K
a
/K
s
values, one be-
tween species and one within species. Under
neutrality, these rates should be equal, given
constant rates of mutation and substitution. If
the between-species ratio significantly exceeds
the within-species ratio, the null hypothesis
can be rejected, suggesting positive selection
between species. Conversely, a larger within-
species value suggests balancing selection or
else a surplus of maladaptive variants (e.g., re-
cessive disease alleles) under weak negative se-
lection within the species (see section, Detect-
ing Selection at the Microevolutionary Level).
Other Rate-Based Methods
Similar to the MKT, the Hudson-Kreitman-
Aguad
´
e (HKA) test uses both divergence and di-
versity data to compare relative rates of change
(Figure 1c). Specifically, the HKA test exam-
ines the ratios of fixed interspecific differences
(D; i.e., substitutions) to within-species poly-
morphisms (P) across loci (59). The test hinges
on the supposition that, for a neutral site, both
D and P are functions of the site’s mutation
rate, which is assumed to have been roughly
constant at least since the point of species di-
vergence. Using a goodness-of-fit test (e.g., χ
2
),
one can check individual sites for deviation from
the neutral D/P ratio, which allows rejection
of the null hypothesis and therefore can be in-
terpreted as evidence for selection. Relatively
large D/P values indicate either that change
contributing to speciation was accelerated (di-
rectional selection between species) or that di-
versity within the species is reduced (directional
selection within species; see section, Detecting
Selection at the Microevolutionary Level). Rel-
atively small values suggest balancing selection
between species.
One advantage of the HKA approach is that
it can be applied to any genetic region, not
just those that code for proteins. In practice,
however, the rate of neutral evolution in
protein-coding regions is much easier to infer
(i.e., by examining the synonymous substitu-
tion rate). The variability of the mutation rate
across different loci, coupled with a lack of
any a priori understanding of which sites (or,
indeed, what percentage of sites) are neutral,
has historically made application of the HKA
test challenging (140). In recent years, how-
ever, researchers have expanded this approach
in a maximum likelihood framework to allow
more efficient multilocus comparisons (135).
By examining multiple sites, one can derive
the expected neutral D/P ratio for a lineage
while accounting for variation in the mutation
rate.
Other studies have used comparative ge-
nomic data to identify elements in the genome
that are highly conserved between disparate
species but show a significantly accelerated rate
of substitution in a particular species or lin-
eage (14, 100, 102). For example, the gene
HAR1F, a noncoding RNA expressed during
brain development, is highly conserved be-
tween chimpanzees and other vertebrates but
has 40 times more substitutions in humans
than expected under neutrality (101). This ap-
proach has been used to identify several hun-
dred human-specific and primate-specific re-
gions (77). Similar relative-rate methods have
also been employed in understanding bacterial
evolution (116).
Phenotypic Methods
The idea of comparing related species and iden-
tifying striking differences can also be applied
to phenotypes. Traits that are conserved across
many closely related species (and thus likely
to be functional) but show extreme differenti-
ation in just one or a few of these species are
strong candidates for natural selection (110).
This approach has been used recently in com-
parative studies of gene expression (13, 97).
The gene SDR16C5, for example, regulates the
www.annualreviews.org
•
Detecting Natural Selection in Genomic Data 103
Annu. Rev. Genet. 2013.47:97-120. Downloaded from www.annualreviews.org
by Harvard University on 11/26/13. For personal use only.

GE47CH05-Sabeti ARI 29 October 2013 12:12
metabolism of retinol, a form of vitamin A
that is common in tree exudates. Slow lorises
and marmosets, which feed on tree bark, show
highly elevated expression levels of SDR16C5 in
the liver compared with their close evolution-
ary cousins, suggesting selection on regulatory
elements as a preventative measure against vi-
tamin A toxicity (97).
Alleles or traits that repeatedly arise in in-
dependent lineages suggest the action of con-
vergent evolution. This signature has been ob-
served in morphological traits, e.g., the loss
of pelvic structures in stickleback fish (18) and
wing pigmentation patterns in Drosophila (105).
It is also seen in viral and bacterial evolution, in
particular in the emergence of drug resistance
(12, 58).
DETECTING SELECTION AT
THE MICROEVOLUTIONARY
LEVEL
Positive selection causes a beneficial allele to
sweep to high prevalence or fixation (100%
prevalence) rapidly within a population. When
a beneficial allele and surrounding variants on
the same haplotype reach high prevalence to-
gether, it produces a population-wide reduc-
tion in genetic diversity (sometimes referred to
as heterozygosity, polymorphism, or variabil-
ity) surrounding the causal allele (119). This
reduction, which persists until recombination
and mutation restore diversity to the popula-
tion at the selected locus, is t he hallmark of a
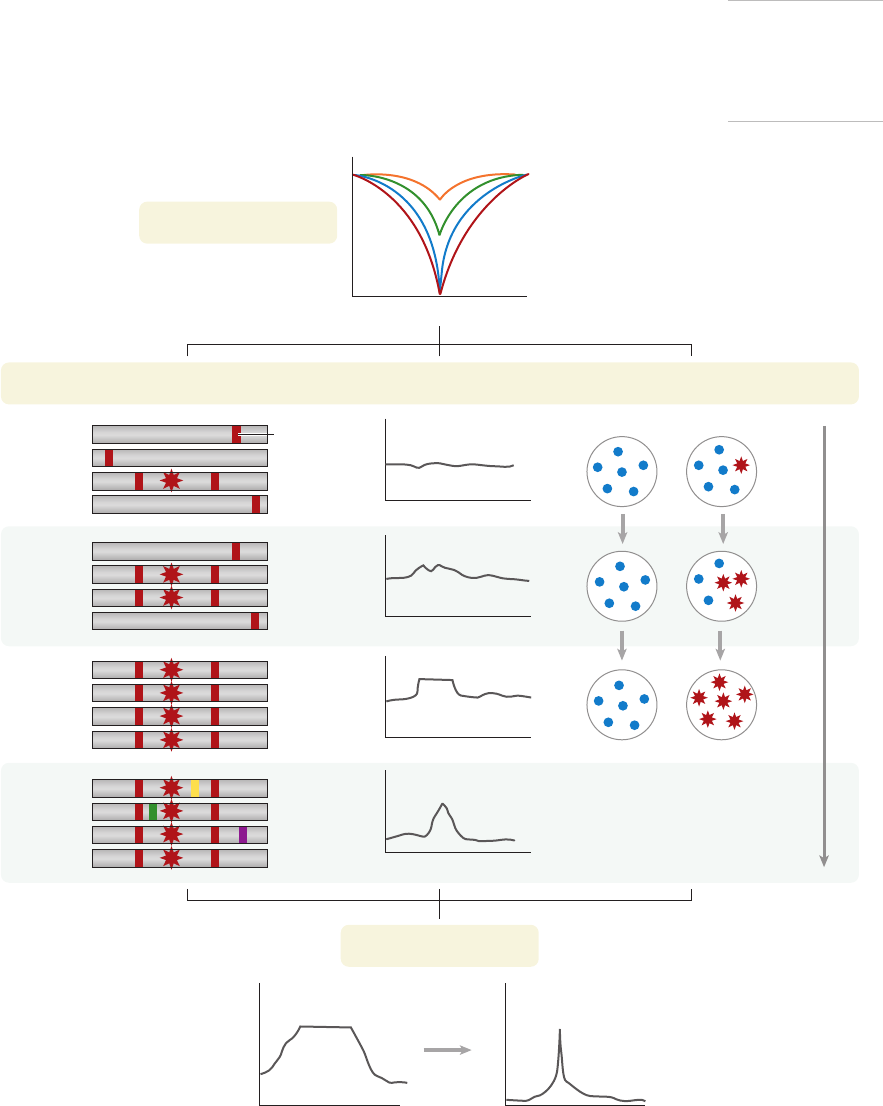
selective sweep (Figure 2a). There are various
ways of quantifying and detecting this signal,
which we discuss in the upcoming two sections.
We then discuss methods based on the envi-
ronmentally specific nature of selection, which
compare populations in which selection is or is
not hypothesized to be at play. We then turn
our attention to methods that combine the re-
sults of multiple tests to provide greater power
and resolution.
Frequency Spectrum–Based Methods
As a selected allele and its nearby hitchhiker
genetic region sweep toward fixation, they shift
the distribution of alleles in the population
(Figure 2b). The sweep causes a population-
wide reduction in the genetic diversity around
the selected locus. New mutations appear on
this homogenous background, but they are
initially rare because they have only recently
appeared in the population. This creates a
surplus of rare alleles (i.e., many sites near
the selected variant have alleles that segregate
at low frequencies). Although the frequency
spectrum shifts back to baseline over time, the
distortion persists for thousands of generations
(several hundred t housand years in the case of
humans). Tajima’s D was the first, and is the
most commonly used, test to detect this signal
(122).
Tajima’s D quantifies this phenomenon by
comparing the number of pair-wise differences
between individuals with the total number
of segregating polymorphisms. Because low-
frequency alleles contribute less to the number
of pair-wise differences in a sample set than do
alleles of moderate frequency, a surplus of rare
−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−→
Figure 2
Methods for detecting selective sweeps at the microevolutionary level. (a) Beneficial mutations bring nearby hitchhiker variants to high
frequency, causing a population-wide reduction in the genetic diversity around the selected locus. This trough in diversity may be
shallower and/or narrower if the sweep is incomplete or if the mutation is not subject to immediate selection (i.e., selection on standing
variation or soft sweep). (b) A beneficial mutation brings nearby derived alleles to high frequency. After the sweep is complete, novel
mutations against a homogenous background create a surplus of rare alleles. (c) A selective sweep causes extended haplotype
homozygosity (EHH), which is a measure of linkage disequilibrium, to rise across the haplotype that contains the selected allele. The
plateau of high EHH begins to break down when novel mutations and recombination gradually restore diversity to the population.
(d ) Differences in allele frequencies, reflecting the population-specific action of selection, cause Wright’s fixation index (F
st
) between
two populations to increase. (e) Composite methods that integrate information from multiple signals of selection can provide finer
resolution and help pinpoint causal variants.
104 Vitti
·
Grossman
·
Sabeti
Annu. Rev. Genet. 2013.47:97-120. Downloaded from www.annualreviews.org
by Harvard University on 11/26/13. For personal use only.
GE47CH05-Sabeti ARI 29 October 2013 12:12
Ancestral: an allele
that was pre-existing
in a population and
from which a derived
allele may arise
alleles inflates the latter value disproportion-
ately to the former value. Thus, smaller (i.e.,
more negative) values of D suggest a surplus of
rare alleles, which may be indicative of positive
selection or population expansion (see section,
Challenges in Applying Statistical Tests for Se-
lection). Several variations on t his method have
been developed to take into account the polarity
of each allele (i.e., which one is derived or ances-
tral based on comparisons with an evolutionary
a Population diversity
Genomic position
Complete sweep de novo
Complete sweep from standing variation
Incomplete sweep
Soft sweep
d Population dierentiation
c Linkage disequilibriumb Frequency spectrum
e Composite methods
P(selection)
Genomic position
Genomic position
TIME
Population A Population B
F
st
~0.5
F
st
~1
Small
F
st
(~0)
EHH
EHH
EHH
EHH
Genomic position
Genomic position
Genomic position
Genomic position
Reaches
xation
High-
frequency
derived
alleles
Diversity
returns
Surplus
of rare
alleles
Rises in
frequency
Novel
variant
Derived
allele
www.annualreviews.org
•
Detecting Natural Selection in Genomic Data 105
Annu. Rev. Genet. 2013.47:97-120. Downloaded from www.annualreviews.org
by Harvard University on 11/26/13. For personal use only.

GE47CH05-Sabeti ARI 29 October 2013 12:12
Derived: an allele
that arises via a novel
mutation and does not
achieve fixation in a
population (as
contrasted with an
ancestral allele)
Genetic drift: change
in allele frequencies
over time due to
chance (e.g. random
sampling)
Linkage
disequilibrium (LD):
tendency of certain
variants on the same
chromosome to be
coinherited at above
chance rates within a
population (e.g., owing
to selection or founder
effects)
outgroup) and to measure the abundance of rare
alleles in different ways (38, 39).
Selective sweeps also distort the frequency
spectrum by increasing the frequency of
derived alleles. Under genetic drift, it takes
many generations to bring neutral mutations
to moderate or high prevalence. However, in a
selective sweep, any derived alleles that reside
near the causal allele also hitchhike to high fre-
quency. Using a similar approach as Tajima’s
D, Fay & Wu’s (34) H compares the number
of pair-wise differences between individuals
to the number of individuals homozygous for
the derived allele. Small values of H indicate
an excess of high-frequency derived alleles,
suggestive of positive selection in the region
examined.
Site frequency spectrum analysis can also be
very useful for other modes of selection, such
as balancing selection, in which an excess of
intermediate-frequency alleles distorts metrics
like Tajima’s D (20). Andr
´
es et al. (5) sought ev-
idence for long-term balancing selection in the
human genome by leveraging frequency spec-
trum methods together with a modification of
the HKA test to detect an excess of diversity in
regions linked to the selected variants. Long-
term balancing selection results in greater co-
alescence times than expected under neutrality
and thus fewer rare alleles.
Linkage Disequilibrium–Based
Methods
As it sweeps through the population, a selected
allele persists in strong linkage disequilibrium
(LD) with its neighboring hitchhiker variants
until recombination causes these associations
to break down. Together, the causal allele and
its linked neighbor variants define a haplotype.
Thus, a third suite of methods for detecting
positive selection looks for extended regions of
strong LD (or, equivalently, long haplotypes)
relative to their prevalence within a population
(Figure 2c). The thought is that such regions
must have swept to high prevalence quickly, or
else recombination would have caused LD to
break down and the haplotype to shorten.
LD-based approaches are particularly useful
for identifying variants that have undergone
a partial or incomplete selective sweep (see
section, Selection on Standing Variation and
Soft Sweeps), in which a new mutation has
risen to a modest frequency in the population
rather than reaching fixation. This is useful
in many species, including humans, as most
novel alleles s ince the out-of-Africa migration
with realistic selection coefficients are unlikely
to have yet reached fixation (Figure 3). For
example, the causal allele of lactase persistence
in Europeans, which has a dominant effect, is
expected to take roughly 50,000 years to reach
Dominant
Recessive
Additive
s = 0.039
Time (ky)
1.0
12.5 25 50
37.5
0.8
0.6
0.4
0.2
0.0
a
Out-of-Africa
Time (ky)
25 50 75 100
s = 0.01
b
Allele frequency
s = 0.005
Time (ky)
25 50 75 100
c
Out-of-Africa
Figure 3
Trajectories of beneficial alleles with realistic selection coefficients simulated in human populations. The fate of a beneficial allele
depends on many factors, including the strength of selection and the extent of the allele’s phenotypic influence (i.e., whether it is
dominant, recessive, etc.). Most alleles with realistic selection coefficients that have arisen since the out-of-Africa migration are
expected to have not yet reached fixation in their respective populations.
106 Vitti
·
Grossman
·
Sabeti
Annu. Rev. Genet. 2013.47:97-120. Downloaded from www.annualreviews.org
by Harvard University on 11/26/13. For personal use only.

GE47CH05-Sabeti ARI 29 October 2013 12:12
Single nucleotide
polymorphism
(SNP): individual
base-pair sites in the
genome of an
organism where
multiple variants exist
fixation, far longer than it has been in existence.
Thus, despite offering one the strongest known
selective advantages (with a selection coeffi-
cient estimated at 0.039) in humans, the allele
frequency is only 80% in Europeans (125).
Beneficial mutations that arose more recently
or were under less extreme selective pressure
are even more likely to remain polymorphic in
the selected population, and many will never
reach fixation because selective pressures can
change greatly over tens of thousands of years.
LD-based approaches can also be used to iden-
tify short-term balancing selection, where the
signal is comparable with that of an incomplete
sweep. For example, a number of papers have
demonstrated long-haplotype signals at the
sickle cell mutation in West Africa (51, 52).
One suite of widely used LD-based tests f or
selection centers around the extended haplo-
type homozygosity statistic (111). One defines
extended haplotype homozygosity (EHH) from
a core region (e.g., a putatively selected allele)
to a specified distance out in both directions
and calculates the probability that any two ran-
domly chosen chromosomes within the pop-
ulation carrying the core region are identical
by descent for the entire region. Thus, as one
travels further from the core region, EHH de-
creases, reflecting the action of recombination
whittling down the haplotype within the pop-
ulation. The long-range haplotype (LRH) test
compares a haplotype’s frequency to its relative
EHH at various distances, looking for haplo-
types that are extended as well as common, sug-
gesting that they rose to high prevalence quickly
enough that recombination has not had time to
break down the haplotype. Zhang et al. (141)
adapted this test by focusing on derived alle-
les (which are believed to be more likely candi-
dates for selective sweeps) as well as by intro-
ducing a genome-wide score. Hanchard et al.
(52) provided the long-range haplotype similar-
ity test, which utilizes a sliding window analysis
to quantify the population-wide homogeneity
of haplotypes.
The integrated haplotype score (iHS) (131)
is an influential variation on EHH. This statis-
tic compares the area under the curve defined
by EHH for the derived and ancestral variants
as one travels further in genetic distance from
the core region. By calculating the area under
the curve defined by EHH, this test captures
the intuition that both extreme EHH for a
short distance and moderate EHH for a longer
distance are suggestive of positive selection.
Another variation is the cross-population
extended haplotype homozygosity (XP-EHH)
statistic (113). This method compares haplo-
type lengths between populations to control
for local variation in recombination rates.
These two methods are complementary in
terms of their scope: Whereas iHS has more
power to detect incomplete sweeps, XP-EHH
is useful when the sweep is near fixation within
one population (99).
Other LD-based tests include the LD decay
(LDD) test, which circumvents the need to de-
termine haplotypes (i.e., by phasing) by limiting
its scope to homozygous single nucleotide poly-
morphism (SNP) sites and inferring the fraction
of recombinant chromosomes at adjacent poly-
morphisms (132). Recently, Wiener & Pong-
Wong (134) developed a new test that fits a re-
gression to heterozygosity data as a function of
genomic position: Selection is inferred on the
basis of the goodness-of-fit to the reduction in
heterozygosity as predicted in a selective sweep.
The strength of this test is that whereas tradi-
tional LD-based approaches are designed for
analysis of SNP data, their regression test can
be used with any genetic marker.
In recent years, a number of researchers
have adapted identity-by-descent (IBD) anal-
yses to selection mapping, invoking essentially
the same conceptual motivations as earlier
EHH-based approaches (15, 50). IBD analyses,
which have been employed in a number of
population history analyses (142), search for
regions in which a set of individuals share a
long stretch of DNA, a pattern that presumably
can only be due to shared ancestry. Although
IBD- and EHH-based methods look for the
same pattern in genomic data, differences
in their computational implementation give
IBD-based approaches the advantage of being
able to detect selection on standing variation
www.annualreviews.org
•
Detecting Natural Selection in Genomic Data 107
Annu. Rev. Genet. 2013.47:97-120. Downloaded from www.annualreviews.org
by Harvard University on 11/26/13. For personal use only.
GE47CH05-Sabeti ARI 29 October 2013 12:12
(see section, Selection on Standing Variation
and Soft Sweeps) with greater power than
EHH-based approaches (3).
Population Differentiation–Based
Methods
An allele’s selective valence is dependent on
the particular environment in which it exists.
Different populations are subject to different
environmental pressures, and as a result, the
traits that would be adaptive in each may be
different. If selection is acting on a locus within
one population but not within other related
populations, then the allele frequencies at that
locus among the populations can differ signifi-
cantly (Figure 2d ). This principle is the foun-
dation of a set of tests that rely on population
differentiation to detect evidence of selection.
The most commonly used metric for popu-
lation differentiation is Wright’s fixation index
(F
st
), which compares the variance of allele
frequencies within and between populations
(57). Comparatively large values of F
st
at a locus
(i.e., relative to neutral regions) indicate stark
differentiation between populations, which is
suggestive of directional selection. Compara-
tively small values indicate that the populations
being compared are homogenous, which may
be indicative of balancing or directional selec-
tion in both. Unlike other methods, population
differentiation–based approaches can detect
many types of selection, including classic
sweeps, sweeps on standing variants, and nega-
tive selection. In recent years, a n umber of alter-
native statistics and variations on F
st
have also
been proposed (for review, see Reference 79).
F
st
-based tests for selection have a long his-
tory, originating with the Lewontin-Krakauer
test (LKT) in 1973 (73). This method uses the
(then limited) available data to estimate F
st
at
multiple loci within n populations and evaluates
the neutrality of this distribution on the basis
of its goodness-of-fit to a χ
2
distribution with
n − 1 degrees of freedom or on the comparison
of this distribution’s variance with a theoretical
predicted value. The production of large
genetic data sets in recent years has made fea-
sible a more robust application of this test, in
which researchers compare the genome-wide
distribution of F
st
to individual loci (2).
Although such outlier approaches are
believed to mitigate the confounding effect
of demographic events—operating on the
understanding that such events affect the
genome in its totality, whereas selection acts
in a locus-specific manner—certain patterns of
migration and mutation within subpopulations
can still produce false positives (82). To correct
for these effects, new variations on this test have
also been developed that incorporate explicit,
user-specified assumptions about demographic
history (11, 31, 129). Bonhomme et al.’s (11)
T
FLK
statistic, for example, modifies the LKT
(labeled T
LK
by the authors) to incorporate a
kinship matrix (F) derived from prespecified
neutral loci to account for historical population
branching. Another line of development
reinterprets the F
st
metric within a Bayesian
framework, often implemented via Markov
chain Monte Carlo algorithms (9, 36, 109).
These approaches utilize F
st
-based statistics
to estimate the posterior probability of a given
allele being under selection.
Other metrics that derive from F
st
improve
its computational power by incorporating more
data. These data come from either a greater
number of populations or a greater number
of allelic sites. On the one hand, following
the former strategy, the locus-specific branch
length metric (LSBL) uses pair-wise calcula-
tions of F
st
from three or more populations
to isolate population-specific changes in allele
frequency relative to a broader genetic context
(117). On the other hand, the cross-population
composite likelihood ratio (XP-CLR) of allele
frequency differentiation extends F
st
to many
loci (22). This method, which is analogous to
the XP-EHH method discussed above, identi-
fies genetic regions in which changes in allele
frequency over many sites occur too quickly
(as assessed by the size of the affected region,
which would gradually return to a neutral
distribution over time) to be due to genetic
drift. More recently, Fariello et al. (32) intro-
duced a new statistic, hapFLK, that examines
108 Vitti
·
Grossman
·
Sabeti
Annu. Rev. Genet. 2013.47:97-120. Downloaded from www.annualreviews.org
by Harvard University on 11/26/13. For personal use only.

GE47CH05-Sabeti ARI 29 October 2013 12:12
Unfolded site
frequency spectrum:
spectrum of allele
frequencies that takes
into account only
derived and not
ancestral alleles
differentiation among populations on the basis
of haplotypes rather than individual alleles.
Composite Methods
As the above discussion suggests, natural se-
lection leaves a number of footprints on the
genome, and each test is designed to pick up
on a slightly different signal. Accordingly, re-
searchers sometimes combine multiple metrics
into composite tests toward the goal of pro-
viding greater power and/or spatial resolution.
These tests come in two distinct forms, both of
which are typically referred to as composite.
First, some methods form a composite score
for a genetic region rather than a single genetic
marker by combining individual scores at all the
markers within the region. The motivation for
such an approach is that, although false pos-
itives may occur at any one site by chance, a
contiguous region of positive markers is much
more likely to represent a bona fide signal (16).
Indeed, because selective sweeps affect whole
haplotypes, one assumes that the signal of selec-
tion extends across a region. Thus, composite
methods that incorporate the same test across
multiple sites improve power and reduce the
false discovery rate. Several of the previously
discussed tests, including iHS, XP-EHH, and
XP-CLR, employ such window-based analyses.
One exemplar of this approach is Kim
& Stephan’s (68) CLR test, which evaluates
the probability of a selective event being
responsible for a surplus of derived alleles (i.e.,
a skew of the unfolded site frequency spectrum)
across multiple sites. Subsequent variations
also incorporated LD-based data (67) and a
goodness-of-fit test to help distinguish selec-
tion from demographic events (63). These tests
calculate a composite likelihood by multiplying
marginal likelihoods for each site considered
within a sequence, and then compare the
composite likelihood under a model in which
a sweep has occurred with t he composite like-
lihood under a model in which no sweep has
occurred. In the above tests, the null hypothesis
was calculated on the basis of a population
genetics model, which Nielsen et al. (89) fur-
ther modified by deriving the null hypothesis
from background patterns of variation in the
data itself. In a later, separate composite test,
Nielsen et al. (87) created a two-dimensional
site frequency spectrum using allele frequen-
cies from two populations; analysis of this
table involved the combination of population
differentiation–based signatures (i.e., F
st
) with
measures for high-frequency derived alleles
and excesses of low-frequency alleles.
Whereas these methods combine the results
of one or a few tests for many variants, other
composite methods combine the results of
many tests at a single site. The purpose of these
composite methods is to utilize complementary
information from different signals in order to
provide better spatial resolution (Figure 2e).
One such line of composite test develop-
ment began with Zeng et al.’s (138) unification
of Tajima’s D and Fay & Wu’s H, each of which
is sensitive to different demographic processes.
Zeng et al. later observed that by limiting them-
selves to site frequency spectrum–based meth-
ods, the power of their test in the presence of
high recombination rates was also limited, and
they opted to further incorporate the Ewens-
Watterson test, which compares the popula-
tion’s Hardy-Weinberg homozygosity to that
predicted under a neutral model (30, 133) and is
largely insensitive to recombination (139). An-
other composite test of this sort was developed
by Grossman et al. (45). This test, called the
composite of multiple signals (CMS) test, in-
corporates metrics from all three suites of meth-
ods discussed here. Specifically, CMS inte-
grates F
st
with iHS and XP-EHH as well as two
new site frequency spectra–based tests that the
authors developed: DAF, which tests for de-
rived alleles t hat are at high frequency relative
to other populations, and iHH, which mea-
sures the absolute rather than relative length of
the haplotype.
MORE COMPLEX MODELS
OF SELECTION
Although the sweep model has been a useful
approach for identifying evidence of selection
www.annualreviews.org
•
Detecting Natural Selection in Genomic Data 109
Annu. Rev. Genet. 2013.47:97-120. Downloaded from www.annualreviews.org
by Harvard University on 11/26/13. For personal use only.
GE47CH05-Sabeti ARI 29 October 2013 12:12
in diverse species, many selective events in hu-
mans and other organisms may not adhere to
this model, and devising new tests to identify
different forms of sweeps continues to be an
area of active research (56, 103). In the selec-
tive sweep model, a novel allele at a single locus
immediately confers a fitness benefit. Two ways
to update the model are to delay the fitness ben-
efit and to allow for multiple loci. In the below
sections, we discuss these two possibilities. We
then turn our attention to ways that extant tests
have been modified to identify different targets
of selection.
Selection on Standing Variation
and Soft Sweeps
Because mutations happen randomly and not in
response to specific selective pressures, alleles
may arise at a time when they are not imme-
diately beneficial. Such neutral alleles might
reach a moderate frequency within the pop-
ulation simply as the result of genetic drift. If
environmental pressures later change to make
such a variant beneficial, the scenario is termed
“selection on standing variation.” Notably, a
standing variant in the EDA signaling pathway
present in seawater fish has been shown to
be under positive selection in freshwater
stickleback fish. The variant, which is largely
hidden in the heterozygous state in seawater
populations, has emerged to cause loss of scales
in multiple distinct freshwater populations (24).
Selection on standing variation is likely to
occur in two scenarios: when the selection co-
efficient and mutation rate are both high and
when the selection coefficient is weak (93). This
latter possibility suggests a potential applica-
tion to complex organisms, such as humans in
particular. Selection on standing variation af-
fects the genome in ways that are compara-
ble to selection on novel variants (8) but can
be more subtle and therefore more difficult to
detect. For example, LD between the stand-
ing variant and its neighbors persists as in a
classical (or hard) sweep; however, compared
with a hard sweep, the resulting trough in di-
versity is shallower, owing to the fact that the
standing variant has time to recombine and as-
sociate with different haplotype backgrounds
(106) (Figure 2a). This fact also distorts the fre-
quency spectrum in a distinctive manner: Com-
pared with a hard sweep, selection on stand-
ing variation creates a greater number of linked
neutral sites that have alleles at intermediate
frequency (106). As the distinction between sig-
natures of hard sweeps and selection on stand-
ing variation may be subtle, Peter et al. (98) offer
an approximate Bayesian computation (ABC)
framework for distinguishing standing variants
from de novo mutations.
A special instance of selection on standing
variation occurs when the standing variant
(or another allele that performs the same
biological function) appears on multiple dis-
tinguishable haplotype backgrounds, e.g., as a
result of recurrent mutation or migration. This
phenomenon is called a soft sweep (55, 93, 94).
Although the term soft sweep is sometimes
mistakenly used to indicate selection on stand-
ing variation more broadly, the two should
be distinguished, as the selective signature
that these trends leave, and consequently the
methods developed to detect them, differ (104).
Through computational simulations, Pen-
nings & Hermisson (94) demonstrated that the
signature of a soft sweep should be in many ways
comparable to that of a hard sweep. Although
frequency-based methods do not have predic-
tive power for soft sweeps—owing to the fact
that soft sweeps may involve an arbitrary num-
ber of distinct haplotypes—LD-based methods
are able to detect the signatures of soft sweeps,
albeit with diminished power. Similar to a hard
sweep, the locus under selection is situated at
the bottom of a trough of genetic diversity.
These results suggest that computational
methods to identify soft sweeps are within
reach; it remains for researchers to fine-tune
current LD-based methods to detect them.
Polygenic Networks and
Ecological Methods
All of the methods discussed thus far assume
that selection acts on one or a few sites at a
110 Vitti
·
Grossman
·
Sabeti
Annu. Rev. Genet. 2013.47:97-120. Downloaded from www.annualreviews.org
by Harvard University on 11/26/13. For personal use only.

GE47CH05-Sabeti ARI 29 October 2013 12:12
Copy number
variants (CNVs):
a form of structural
variant in which
multiple copies of a
genetic region exist
Microsatellites:
genetic regions that
consist of repeating
sequences of two to six
base pairs. Also
referred to as short
tandem repeats (STRs)
or simple sequence
repeats (SSRs)
Structural variants
(SVs): alterations in
the genome that affect
relatively large
chromosomal regions,
including deletions
and insertions (indels),
translocations,
inversions, and
duplications
time. However, given the known importance
of polygenic networks and of epistatic interac-
tions, researchers have suggested that selection
may more often act on multiple sites in tandem,
causing coordinated and distributed shifts in al-
lele frequencies (53, 104).
One way to identify polygenic groups of sites
under selection is to incorporate ecological in-
formation. By binning related populations ac-
cording to presumably relevant variables (e.g.,
habitat, climate, mode of sustenance, etc.), one
can seek shifts in allele frequency shared across
ecologically similar populations. Joost et al.
(65) formalized this approach as the spatial
analysis method (SAM), using multiple univari-
ate logistic regressions to test for association
between allele frequencies and environmental
variables. Jones et al. (64) use a similar approach
in their comparison of marine and freshwa-
ter sticklebacks from globally distributed pop-
ulations to identify loci consistently associated
with habitat, and Hancock et al. (53) perform a
similar analysis to identify ecologically relevant
loci in humans.
An important limitation of ecological
approaches is their reliance on user-specified
variables (104). These methods run the risk
of being biased by the information put in or
left out. Polygenic selection can be detected
without the risk of this bias by examining
shared functional sets, such as quantitative trait
loci (QTLs), in which multiple genetic regions
contribute to a single trait. Selection acting on a
network of QTLs can be inferred on the basis of
a significant bias in their directionality, i.e., the
tendency of a locus to either amplify or lessen
the magnitude of the phenotype (91). Although
under neutrality, the distribution of positive or
negative QTLs may be random, an overrepre-
sentation of one or the other type of loci within
a lineage is s uggestive of selection. Fraser et al.
(37) developed a framework in which this test
can be applied in a genome-wide scan, focusing
on regulatory elements [i.e., expression QTLs
(eQTLs)] in mice. Similarly, Simonson et al.
(118) performed a genome-wide scan with
attention to genic networks known to be
involved in an oxygen-carry capacity to reveal
adaptation to high altitudes in a Tibetan
population.
Alternative Targets of Selection
Most natural selection studies to date have fo-
cused on genetic changes at the single nu-
cleotide level, primarily because they have been
the most accessible from a technological stand-
point, through advances in protein analysis and
SNP genotyping. Given their mutation mech-
anism, which typically creates simple biallelic
changes of unique origin, they also can be more
easily incorporated into statistical tests for se-
lection. Moreover, SNPs are useful in such tests
because they can act as markers: Nearby vari-
ants in LD with a S NP can be detected by using
said SNP as a proxy.
Many other genetic alterations that affect
an organism’s phenotype may be subject to
selection, including copy number variants
(CNVs) (115), microsatellites (46), chromo-
somal rearrangements (e.g., indels, inversions,
and translocations) (35), polygenic networks
(discussed above), and epigenetic annotations
(127). One of the first elucidated examples
of selection were CNVs of α-andβ-globin
genes implicated in thalassemia, which, along
with sickle cell anemia, confer resistance to
malaria (6, 137). More recently, increased
CNV counts of the gene for amylase have
also been demonstrated to be associated with
diets containing larger amounts of starch
(96). Another example is a major inversion on
chromosome 17 in humans that was shown to
be associated with greater reproductive success
in an Icelandic population (121) and contains
population genetics evidence of positive selec-
tion. Structural variants (SVs), such as CNVs
and inversions, are often subject to negative
selection (especially those that may cause
frameshifts in protein-coding regions) (76)
or can lead to relaxed evolutionary constraint
through gene duplication (70). The many tests
for selection described above may be applied
to SVs, although the broad diversity of variants
under the umbrella term SV and the large
effects they can have on genomic architecture
www.annualreviews.org
•
Detecting Natural Selection in Genomic Data 111
Annu. Rev. Genet. 2013.47:97-120. Downloaded from www.annualreviews.org
by Harvard University on 11/26/13. For personal use only.

GE47CH05-Sabeti ARI 29 October 2013 12:12
Epigenome:
annotations to the
DNA molecule that
alter patterns of gene
expression but do not
change the sequence
make the systematic detection of selected
variants challenging (61).
The recent discovery that certain epigenetic
arrangements are heritable across many gen-
erations also raises the possibility of selection
acting on the epigenome (62, 108). Such neo-
Lamarckian selection has been detected in or-
chids using SAM (92). It remains to be clari-
fied to what extent such modes of selection are
prevalent, but it is an area of active interest.
CHALLENGES IN APPLYING
STATISTICAL TESTS
FOR SELECTION
Although each approach has its own particular
strengths and limitations, there are a number
of challenges that are shared among these
tests, particularly in the interpretation of
their significance. A neutrality test may allow
rejection of the null hypothesis, but there
are many possible explanations other than
selection for the genomic results observed. For
example, demographic events (e.g., migration,
expansions, and bottlenecks) can often create
selection-mimicking signals. Historically, most
studies have aimed to rule out this possibility by
comparing locus-specific data to genome-wide
data, as demographic events are understood
to affect the genome in its totality, whereas
selection acts in a more targeted manner (17).
In recent years, however, some have questioned
this outlier approach, arguing that if selection
is pervasive (as in Drosophila; see Reference
74), then distributed patterns of genetic hitch-
hiking would be misinterpreted as reflecting
demographic events (47). More generally,
the recognition that the effects of selection
and demography may be interconnected have
led some to adopt other approaches, such as
explicitly estimating demographic parameters,
including population structure, through various
computational frameworks and incorporating
these into subsequent analyses (for examples,
see Reference 31; for review, see Reference
75). Another related issue is that false positives
can be produced when tests implicate neutral
variants in strong LD with a causal allele (124).
Even when these confounding effects can be
ruled out, the interpretation of selection may
not be straightforward. For example, rate-based
tests implicate regions in which evolutionary
change has been accelerated: This may be
due to positive selection of novel variants, but
the relaxation of selective constraint (i.e., of
purifying selection) over a region may have
the same effect. Distinguishing between these
possibilities involves case-by-case analysis. In a
study of the evolution of CNVs in humans, for
example, Nguyen et al. (84) ruled out positive
selection in regions in which they observed an
inverse relationship between rates of change
and rates of recombination. More generally,
however, functional analysis of candidate
regions can help adjudicate between these
two possibilities: If the derived variant has
no potentially fitness-enhancing variation of
function, relative to the ancestral, then the re-
laxation of selective constraint is the more likely
explanation.
Another recurrent challenge for researchers
is accounting for systematic biases that may
be present in genomic data. The majority of
selection studies to date have utilized SNP
data, which is collected using genotyping arrays
designed to detect known polymorphisms.
The practical limitations of SNP discovery
protocols mean that low-frequency alleles
may go undetected, in which case they are
excluded from these arrays. These arrays can
therefore generate data that may be unrepre-
sentative of the full extent of genetic diversity,
a phenomenon known as ascertainment bias
(23). This sampling of the data can artificially
distort allele frequency measures as well as
derivative statistics that include LD. When the
SNP discovery protocol is known, statistical
measures can be taken to counteract the effect
of ascertainment bias (86, 88, 107). In addition,
genotyping assays that incorporate variable in-
tensity oligonucleotide (VINO) probes can be
used to mitigate the number of polymorphisms
overlooked as a result of ascertainment bias
(26).
Another salient issue for researchers in-
vestigating natural selection, particularly for
112 Vitti
·
Grossman
·
Sabeti
Annu. Rev. Genet. 2013.47:97-120. Downloaded from www.annualreviews.org
by Harvard University on 11/26/13. For personal use only.

GE47CH05-Sabeti ARI 29 October 2013 12:12
Table 2 Using selection scans to study human evolution
Gene under
selection
Population(s)
Genomic
evidence for
selection
Functional
evidence
Putative adaptive role References
FOXP2 All (selection predates
out-of-Africa
migration)
Accelerated
evolution in
coding region,
D, H
Mouse transgenic Affects development of
corticobasal ganglia
circuits; thought to be
involved in mechanics of
speech
(28, 29)
LCT Northern Europeans,
East Africans
(pastoralist societies)
EHH, iHS; F
st
analysis
Human association
study; in vitro
lactase expression
assay
Confers lactase
persistence; allows
digestion of lactose into
adulthood
(10, 125)
EDAR East Asians and Native
Americans
CMS Human association
study; mouse
transgenic
Affects morphology of
hair, sweat glands, and
mammary glands
(45, 66)
TLR5 West Africans CMS In vitro assay of
NF-κB pathway
activation
Modulates immune
response to bacterial
flagellin
(44, 45)
DARC African populations in
malaria-endemic
regions
F
st
Human association
study
Heterozygosis reduces
susceptibility to malaria
(49, 66a, 80)
APOL1 African populations in
trypanosome-endemic
regions
CMS In vitro assay of
response to
trypanosome
invasion
Modulates susceptibility
to trypanosomiasis
(45, 95, 128)
HBB African populations in
malaria-endemic
regions
LRH
LRH similarity
Human association
study
Heterozygosis reduces
susceptibility to malaria
(4, 51, 72,
111)
EPAS1,
EGLN,
et al.
Tibetans iHS, XP-EHH Human association
study
Selected variants decrease
hemoglobin
concentration and
modulate hypoxia
response
(41, 118)
SLC24A5,
SLC45A2
Europeans F
st
analysis,
XP-EHH, CMS
Human association
study; in vitro assay
of melanocyte
cultures; zebrafish
transgenic
Decreases melanin
pigmentation in skin
(25, 90, 126)
CBARA1,
VAV3, et al.
Ethiopian-highland
populations
LSBL, iHS,
XP-EHH
Human association
study
Selected variants decrease
hemoglobin
concentration and
modulate hypoxia
response
(114)
Abbreviations: CMS, composite of multiple signals; EHH, extended haplotype homozygosity; F
st
, Wright’s fixation index; iHS, integrated haplotype
score; LRH, long-range haplotype; LSBL, locus-specific branch length metric; XP-EHH, cross-population extended haplotype homozygosity.
www.annualreviews.org
•
Detecting Natural Selection in Genomic Data 113
Annu. Rev. Genet. 2013.47:97-120. Downloaded from www.annualreviews.org
by Harvard University on 11/26/13. For personal use only.

GE47CH05-Sabeti ARI 29 October 2013 12:12
Pleiotropy: atrendin
which one genotype
affects multiple
phenotypes
those studying it in humans, is the potential for
misinterpretation of results and their societal
significance. By attending to linguistic sub-
tleties and employing caution in disseminating
results, researchers can help prevent unethical
application of evolutionary research (130).
FROM GENOME SCANS TO
EVOLUTIONARY HYPOTHESES
The ultimate validation of genomic metrics
of selection is the demonstration that putative
selective variants have phenotypic effects
with import for organismal fitness (Table 2).
Functional understanding of a candidate region
begins with the fine-mapping of that region so
as to localize the signal. Until recently, localiz-
ing signals of selection was a major challenge,
but new composite methods and full-genome
sequence data provide stronger resolution
that can allow researchers to identify tractable
candidates for functional scrutiny (44, 45).
Once individual alleles have been identified for
experimentation, researchers can measure the
effects of said alleles as compared with their
wild-type analogs. Genomic annotation can be
informative for experimental design by sug-
gesting the most probable types of traits that
a variant may affect or by suggesting the types
of cells in which a variant is most commonly
expressed.
Phenotypic screening may then proceed
through an association study of various
traits in the organism in question, although
background genetic variation can introduce
noise into the data. To correct for this,
researchers may instead compare the derived
and ancestral variants against the same genetic
background introduced into a cell line in vitro
or into model organisms in vivo. Even in
such situations, however, the possibility that a
variant has pleiotropic effects makes it difficult
to discern whether a functional follow-up study
correctly identifies the selective significance of
the variant in question (7). Although exhaustive
phenotype screens are not possible, researchers
can bolster the strength of their evidence by
screening through as comprehensive a list of
possible effects as possible. For example, Enard
et al. (28) introduced two human-specific
amino acid substitutions in the FOXP2 gene
into mice and screened approximately three
hundred traits, ultimately finding that only
a small fraction of these (those involving the
structure and function of corticobasal ganglia
circuits) were significantly different between
humanized and wild-type mice.
Creating a case for selection necessitates a
combination of genomic and functional evi-
dence. With the availability of large population
genetics data sets, statistical methods to inter-
pret that data, and increasingly sophisticated
technologies for transgenesis and other func-
tional methods, researchers are moving into a
new era of natural selection studies, in which
both the causes and effects of changes to the
genomes of humans and other organisms can
be modeled and understood.
SUMMARY POINTS
1. The development of genotyping and sequencing technologies has allowed for the full
realization and application of methods to investigate selection on the basis of theory from
the fields of comparative genomics and population genetics.
2. Methods to detect selection in the genome may be categorized by their effective timescale
(i.e., macro- versus microevolutionary) as well as by the types of data they utilize (i.e.,
interspecies divergence data, intraspecies diversity data, or a combination of these), or
the type of selective signature they identify.
114 Vitti
·
Grossman
·
Sabeti
Annu. Rev. Genet. 2013.47:97-120. Downloaded from www.annualreviews.org
by Harvard University on 11/26/13. For personal use only.

GE47CH05-Sabeti ARI 29 October 2013 12:12
3. Tests to detect selection at the macroevolutionary level make interspecific comparisons,
often aided by phylogenetic considerations, of the rates of change at the nucleotide level
and look for genetic regions in species that have experienced accelerated change.
4. Tests for microevolutionary selection come in a broad range of formats but often aim
to detect regions of reduced genetic diversity, which is indicative of a selective sweep.
Other tests compare populations in which selection is or is not hypothesized to be at play
and measure the extent of differentiation between them. Combining multiple tests can
increase power and resolution.
5. An active area of research is the development of tests for modes of selection that do
not adhere to the selective sweep model. Among these are polygenic selection and soft
sweeps.
6. Genomic evidence for selection is suggestive but not conclusive. A combination of ge-
nomic and functional evidence constitutes the current standard for the field.
FUTURE ISSUES
1. How can we make tests for alternative selective modes (soft sweeps, polygenic selection,
etc.) more robust?
2. How can we accurately quantify the prevalence of selection and the relative contribution
of drift in humans and other organisms?
3. Can we develop high-throughput assays for functional analysis and validation of candidate
variants?
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that
might be perceived as affecting the objectivity of this review.
LITERATURE CITED
1. Akey JM. 2009. Constructing genomic maps of positive selection in humans: Where do we go from here?
Genome Res. 19(5):711–22
2. Akey JM, Zhang G, Zhang K, Jin L, Shriver MD. 2002. Interrogating a high-density SNP map for
signatures of natural selection. Genome Res. 12(12):1805–14
3. Albrechtsen A, Moltke I, Nielsen R. 2010. Natural selection and the distribution of identity-by-descent
in the human genome. Genetics 186(1):295–308
4. Allison AC. 1954. Protection afforded by sickle-cell trait against subtertian malarial infection. Br. Med.
J. 1(4857):290–94
5. Andr
´
es AM, Hubisz MJ, Indap A, Torgerson DG, Degenhardt JD, et al. 2009. Targets of balancing
selection in the human genome. Mol. Biol. Evol. 26(12):2755–64
6. Barrai I, Rosito A, Cappellozza G, Cristofori G, Vullo C, et al. 1984. Beta-thalassemia in the Po Delta:
selection, geography, and population structure. Am. J. Hum. Genet. 36(5):1121–34
7. Barrett RDH, Hoekstra HE. 2011. Molecular spandrels: tests of adaptation at the genetic level. Nat. Rev.
Genet. 12(11):767–80
8. Barrett RDH, Schluter D. 2008. Adaptation from standing genetic variation. Trends Ecol. Evol. (Amst.)
23(1):38–44
www.annualreviews.org
•
Detecting Natural Selection in Genomic Data 115
Annu. Rev. Genet. 2013.47:97-120. Downloaded from www.annualreviews.org
by Harvard University on 11/26/13. For personal use only.
GE47CH05-Sabeti ARI 29 October 2013 12:12
9. Beaumont MA, Balding DJ. 2004. Identifying adaptive genetic divergence among populations from
genome scans. Mol. Ecol. 13(4):969–80
10. Bersaglieri T, Sabeti PC, Patterson N, Vanderploeg T, Schaffner SF, et al. 2004. Genetic signatures of
strong recent positive selection at the lactase gene. Am. J. Hum. Genet. 74(6):1111–20
11. Bonhomme M, Chevalet C, Servin B, Boitard S, Abdallah J, et al. 2010. Detecting selection in population
trees: the Lewontin and Krakauer test extended. Genetics 186(1):241–62
12. Boucher CAB, O’Sullivan E, Mulder JW, Ramautarsing C, Kellam P, et al. 1992. Ordered appearance of
zidovudine resistance mutations during treatment of 18 human immunodeficiency virus-positive subjects.
J. Infect. Dis. 165(1):105–10
13. Brawand D, Soumillon M, Necsulea A, Julien P, Cs
´
ardi G, et al. 2011. The evolution of gene expression
levels in mammalian organs. Nature 478(7369):343–48
14. Burbano HA, Green RE, Maricic T, Lalueza-Fox C, De la Rasilla M, et al. 2012. Analysis of human
accelerated DNA regions using archaic hominin genomes. PLoS ONE 7(3):e32877
15. Cai Z, Camp NJ, Cannon-Albright L, Thomas A. 2011. Identification of regions of positive selection
using shared genomic segment analysis. Eur. J. Hum. Genet. 19(6):667–71
16. Carlson CS, Thomas DJ, Eberle MA, Swanson JE, Livingston RJ, et al. 2005. Genomic regions exhibiting
positive selection identified from dense genotype data. Genome Res. 15(11):1553–65
17. Cavalli-Sforza LL. 1966. Population structure and human evolution. Proc. R. Soc. Lond. Ser. B.
164(995):362–79
18. Chan YF, Marks ME, Jones FC, Villarreal G, Shapiro MD, et al. 2010. Adaptive evolution of pelvic
reduction in sticklebacks by recurrent deletion of a Pitx1 enhancer. Science 327(5963):302–5
19. Charlesworth B, Morgan MT, Charlesworth D. 1993. The effect of deleterious mutations on neutral
molecular variation. Genetics 134(4):1289–303
20. Charlesworth D. 2006. Balancing selection and its effects on sequences in nearby genome regions. PLoS
Genet. 2(4):e64
21. Chamary JV, Parmley JL, Hurst LD. 2006. Hearing silence: non-neutral evolution at synonymous sites
in mammals. Nat. Rev. Genet. 7(2):98–108
22. Chen H, Patterson N, Reich D. 2010. Population differentiation as a test for selective sweeps. Genome
Res. 20(3):393–402
23. Clark AG, Hubisz MJ, Bustamante CD, Williamson SH, Nielsen R. 2005. Ascertainment bias in studies
of human genome-wide polymorphism. Genome Res. 15(11):1496–502
24. Colosimo PF, Hosemann KE, Balabhadra S, Villarreal G Jr, Dickson M, et al. 2005. Widespread parallel
evolution in sticklebacks by repeated fixation of ectodysplasin alleles. Science 307(5717):1928–33
25. Cook AL, Chen W, Thurber AE, Smit DJ, Smith AG, et al. 2009. Analysis of cultured human melanocytes
based on polymorphisms within the SLC45A2/MATP, SLC24A5/NCKX5, and OCA2/P loci. J. Investig.
Dermatol. 129(2):392–405
26. Didion JP, Yang H, Sheppard K, Fu C-P, McMillan L, et al. 2012. Discovery of novel variants in
genotyping arrays improves genotype retention and reduces ascertainment bias. BMC Genomics 13:34
27. Egea R, Casillas S, Barbadilla A. 2008. Standard and generalized McDonald-Kreitman test: a website to
detect selection by comparing different classes of DNA sites. Nucleic Acids Res. 36:W157–62
28. Enard W, Gehre S, Hammerschmidt K, H
¨
olter SM, Blass T, et al. 2009. A humanized version of Foxp2
affects cortico-basal ganglia circuits in mice. Cell 137(5):961–71
29. Enard W, Przeworski M, Fisher SE, Lai CSL, Wiebe V, et al. 2002. Molecular evolution of FOXP2,a
gene involved in speech and language. Nature 418(6900):869–72
30. Ewens WJ. 1972. The sampling theory of selectively neutral alleles. Theor. Popul. Biol. 3(1):87–112
31. Excoffier L, Hofer T, Foll M. 2009. Detecting loci under selection in a hierarchically structured popu-
lation. Heredity (Edinb.) 103(4):285–98
32. Fariello MI, Boitard S, Naya H, San Cristobal M, Servin B. 2013. Detecting signatures of selection
through haplotype differentiation among hierarchically structured populations.
Genetics 193:929–41
33. Fay JC. 2011. Weighing the evidence for adaptation at the molecular level. Trends Genet. 27(9):343–49
34. Fay JC, Wu CI. 2000. Hitchhiking under positive Darwinian selection. Genetics 155(3):1405–13
35. Feuk L, Carson AR, Scherer SW. 2006. Structural variation in the human genome. Nat. Rev. Genet.
7(2):85–97
116 Vitti
·
Grossman
·
Sabeti
Annu. Rev. Genet. 2013.47:97-120. Downloaded from www.annualreviews.org
by Harvard University on 11/26/13. For personal use only.
GE47CH05-Sabeti ARI 29 October 2013 12:12
36. Foll M, Gaggiotti O. 2008. A genome-scan method to identify selected loci appropriate for both dominant
and codominant markers: a Bayesian perspective. Genetics 180(2):977–93
37. Fraser HB, Babak T, Tsang J, Zhou Y, Zhang B, et al. 2011. Systematic detection of polygenic cis-
regulatory evolution. PLoS Genet. 7(3):e1002023
38. Fu Y-X. 1997. Statistical tests of neutrality of mutations against population growth, hitchhiking and
background selection. Genetics 147(2):915–25
39. Fu YX, Li WH. 1993. Statistical tests of neutrality of mutations. Genetics 133(3):693–709
40. Fumagalli M, Sironi M, Pozzoli U, Ferrer-Admettla A, Pattini L, Nielsen R. 2011. Signatures of environ-
mental genetic adaptation pinpoint pathogens as the main selective pressure through human evolution.
PLoS Genet. 7(11):e1002355
41. Ge R-L, Simonson TS, Cooksey RC, Tanna U, Qin G, et al. 2012. Metabolic insight into mechanisms
of high-altitude adaptation in Tibetans. Mol. Genet. Metab. 106(2):244–47
42. Gould S. 1978. Sociobiology: the art of storytelling. New Sci. 80(1129):530–33
43. Graur D, Li W-H. 2000. Fundamentals of Molecular Evolution. Sunderland, MA: Sinauer Assoc.
44. Grossman SR, Andersen KG, Shlyakhter I, Tabrizi S, Winnicki S, et al. 2013. Identifying recent adap-
tations in large-scale genomic data. Cell 152(4):703–13
45. Grossman SR, Shylakhter I, Karlsson EK, Byrne EH, Morales S, et al. 2010. A composite of multiple
signals distinguishes causal variants in regions of positive selection. Science 327(5967):883–86
46. Haasl RJ, Payseur BA. 2012. Microsatellites as targets of natural selection. Mol. Biol. Evol. 30(2):285–98
47. Hahn MW. 2008. Toward a selection theory of molecular evolution. Evolution 62(2):255–65
48. Haldane JBS. 2006. Disease and evolution. In Malaria: Genetic and Evolutionary Aspects,ed.KR
Dronamraju, P Arese, pp. 175–87. New York: Springer
49. Hamblin MT, DiRienzo A. 2000. Detection of the signature of natural selection in humans: evidence
from the Duffy blood group locus. Am. J. Hum. Genet. 66(5):1669–79
50. Han L, Abney M. 2012. Using identity by descent estimation with dense genotype data to detect positive
selection. Eur. J. Hum. Genet. 21(2):205–11
51. Hanchard N, Elzein A, Trafford C, Rockett K, Pinder M, et al. 2007. Classical sickle β-globin haplotypes
exhibit a high degree of long-range haplotype similarity in African and Afro-Caribbean populations. BMC
Genet. 8(1):52
52. Hanchard NA, Rockett KA, Spencer C, Coop G, Pinder M, et al. 2006. Screening for recently selected
alleles by analysis of human haplotype similarity. Am. J. Hum. Genet. 78(1):153–59
53. Hancock AM, Witonsky DB, Ehler E, Alkorta-Aranburu G, Beall C, et al. 2010. Colloquium paper:
human adaptations to diet, subsistence, and ecoregion are due to subtle shifts in allele frequency. Proc.
Natl. Acad. Sci. USA 107(Suppl. 2):8924–30
54. Hancock AM, Witonsky DB, Gordon AS, Eshel G, Pritchard JK, et al. 2008. Adaptations to climate in
candidate genes for common metabolic disorders. PLoS Genet. 4(2):e32
55. Hermisson J, Pennings PS. 2005. Soft sweeps: molecular population genetics of adaptation from standing
genetic variation. Genetics 169(4):2335–52
56. Hernandez RD, Kelley JL, Elyashiv E, Melton SC, Auton A, et al. 2011. Classic selective sweeps were
rare in recent human evolution. Science 331(6019):920–24
57. Holsinger KE, Weir BS. 2009. Genetics in geographically structured populations: defining, estimating
and interpreting F(ST). Nat. Rev. Genet. 10(9):639–50
58. Holt KE, Parkhill J, Mazzoni CJ, Roumagnac P, Weill F-X, et al. 2008. High-throughput sequencing
provides insights into genome variation and evolution in Salmonella Typhi. Nat. Genet. 40(8):987–93
59. Hudson RR, Kreitman M, Aguad
´
e M. 1987. A test of neutral molecular evolution based on nucleotide
data. Genetics 116(1):153–59
60. Hurst LD. 2002. The Ka/Ks ratio: diagnosing the form of sequence evolution. Trends Genet. 18(9):486
61. Iskow RC, Gokcumen O, Lee C. 2012. Exploring the role of copy number variants in human adaptation.
Trends Genet. 28(6):245–57
62. Jablonka E, Raz G. 2009. Transgenerational epigenetic inheritance: prevalence, mechanisms, and im-
plications for the study of heredity and evolution. Q. Rev. Biol. 84(2):131–76
63. Jensen JD, Kim Y, DuMont VB, Aquadro CF, Bustamante CD. 2005. Distinguishing between selective
sweeps and demography using DNA polymorphism data. Genetics 170(3):1401–10
www.annualreviews.org
•
Detecting Natural Selection in Genomic Data 117
Annu. Rev. Genet. 2013.47:97-120. Downloaded from www.annualreviews.org
by Harvard University on 11/26/13. For personal use only.
GE47CH05-Sabeti ARI 29 October 2013 12:12
64. Jones FC, Grabherr MG, Chan YF, Russell P, Mauceli E, et al. 2012. The genomic basis of adaptive
evolution in threespine sticklebacks. Nature 484(7392):55–61
65. Joost S, Bonin A, Bruford MW, Despr
´
es L, Conord C, et al. 2007. A spatial analysis method (SAM)
to detect candidate loci for selection: towards a landscape genomics approach to adaptation. Mol. Ecol.
16(18):3955–69
66. Kamberov YG, Wang S, Tan J, Gerbault P, Wark A, et al. 2013. Modeling recent human evolution in
mice by expression of a selected EDAR variant. Cell 152(4):691–702
66a. Kasehagen LJ, Mueller I, Kinboro B, Bockarie MJ, Reeder JC, et al. 2007. Reduced Plasmodium vivax
erythrocyte infection in PNG Duffy-negative heterozygotes. PLoS ONE 2(3):e336
67. Kim Y, Nielsen R. 2004. Linkage disequilibrium as a signature of selective sweeps. Genetics 167(3):1513–
24
68. Kim Y, Stephan W. 2002. Detecting a local signature of genetic hitchhiking along a recombining chro-
mosome. Genetics 160(2):765–77
69. Kimura M. 1985. The Neutral Theory of Molecular Evolution. Cambridge: Cambridge Univ. Press
70. Kondrashov FA. 2012. Gene duplication as a mechanism of genomic adaptation to a changing environ-
ment. Proc. R. Soc. Lond. Ser. B 279(1749):5048–57
71. Kreitman M, Akashi H. 1995. Molecular evidence for natural selection. Annu. Rev. Ecol. Syst. 26:403–22
72. Kwiatkowski DP. 2005. How malaria has affected the human genome and what human genetics can teach
us about malaria. Am. J. Hum. Genet. 77(2):171–92
73. Lewontin RC, Krakauer J. 1973. Distribution of gene frequency as a test of the theory of the selective
neutrality of polymorphisms. Genetics 74(1):175–95
74. Li H, Stephan W. 2006. Inferring the demographic history and rate of adaptive substitution in Drosophila.
PLoS Genet. 2(10):e166
75. Li J, Li H, Jakobsson M, Li S, Sj
¨
odin P, Lascoux M. 2012. Joint analysis of demography and selection
in population genetics: Where do we stand and where could we go? Mol. Ecol. 21(1):28–44
76. Li Y, Zheng H, Luo R, Wu H, Zhu H, et al. 2011. Structural variation in two human genomes mapped
at single-nucleotide resolution by whole genome de novo assembly. Nat. Biotechnol. 29(8):723–30
77. Lindblad-Toh K, Garber M, Zuk O, Lin MF, Parker BJ, et al. 2011. A high-resolution map of human
evolutionary constraint using 29 mammals. Nature 478(7370):476–82
78. McDonald JH, Kreitman M. 1991. Adaptive protein evolution at the Adh locus in Drosophila. Nature
351(6328):652–54
79. Meirmans PG, Hedrick PW. 2011. Assessing population structure: FST and related measures. Mol. Ecol.
Resour. 11(1):5–18
80. Miller LH, Mason SJ, Clyde DF, McGinniss MH. 1976. The resistance factor to Plasmodium vivax in
blacks. N. Engl. J. Med. 295(6):302–4
81. Mullen LM, Vignieri SN, Gore JA, Hoekstra HE. 2009. Adaptive basis of geographic variation:
genetic, phenotypic and environmental differences among beach mouse populations. Proc. Biol. Sci.
276(1674):3809–18
82. Nei M, Maruyama T. 1975. Letters to the editors: Lewontin-Krakauer test for neutral genes. Genetics
80(2):395
83. Nei M, Suzuki Y, Nozawa M. 2010. The neutral theory of molecular evolution in the genomic era. Annu.
Rev. Genomics Hum. Genet. 11:265–89
84. Nguyen D-Q, Webber C, Hehir-Kwa J, Pfundt R, Veltman J, Ponting CP. 2008. Reduced purify-
ing selection prevails over positive selection in human copy number variant evolution. Genome Res.
18(11):1711–23
85. Nielsen R. 2005. Molecular signatures of natural s election. Annu. Rev. Genet. 39:197–218
86. Nielsen R, Hubisz MJ, Clark AG. 2004. Reconstituting the frequency spectrum of ascertained single-
nucleotide polymorphism data.
Genetics 168(4):2373–82
87. Nielsen R, Hubisz MJ, Hellmann I, Torgerson D, Andr
´
es AM, et al. 2009. Darwinian and demographic
forces affecting human protein coding genes. Genome Res. 19(5):838–49
88. Nielsen R, Signorovitch J. 2003. Correcting for ascertainment biases when analyzing SNP data: appli-
cations to the estimation of linkage disequilibrium. Theor. Popul. Biol. 63(3):245–55
118 Vitti
·
Grossman
·
Sabeti
Annu. Rev. Genet. 2013.47:97-120. Downloaded from www.annualreviews.org
by Harvard University on 11/26/13. For personal use only.
GE47CH05-Sabeti ARI 29 October 2013 12:12
89. Nielsen R, Williamson S, Kim Y, Hubisz MJ, Clark AG, Bustamante C. 2005. Genomic scans for selective
sweeps using SNP data. Genome Res. 15(11):1566–75
90. Norton HL, Kittles RA, Parra E, McKeigue P, Mao X, et al. 2007. Genetic evidence for the convergent
evolution of light skin in Europeans and East Asians. Mol. Biol. Evol. 24(3):710–22
91. Orr HA. 1998. Testing natural selection versus genetic drift in phenotypic evolution using quantitative
trait locus data. Genetics 149(4):2099–104
92. Paun O, Bateman RM, Fay MF, Hedr
´
en M, Civeyrel L, Chase MW. 2010. Stable epigenetic effects
impact adaptation in allopolyploid orchids (Dactylorhiza: Orchidaceae). Mol. Biol. Evol. 27(11):2465–73
93. Pennings PS, Hermisson J. 2006. Soft sweeps II: molecular population genetics of adaptation from
recurrent mutation or migration. Mol. Biol. Evol. 23(5):1076–84
94. Pennings PS, Hermisson J. 2006. Soft sweeps III: the signature of positive selection from recurrent
mutation. PLoS Genet. 2(12):e186
95. P
´
erez-Morga D, Vanhollebeke B, Paturiaux-Hanocq F, Nolan DP, Lins L, et al. 2005. Apolipoprotein
L-I promotes trypanosome lysis by forming pores in lysosomal membranes. Science 309(5733):469–72
96. Perry GH, Dominy NJ, Claw KG, Lee AS, Fiegler H, et al. 2007. Diet and the evolution of human
amylase gene copy number variation. Nat. Genet. 39(10):1256–60
97. Perry GH, Melsted P, Marioni JC, Wang Y, Bainer R, et al. 2012. Comparative RNA sequencing reveals
substantial genetic variation in endangered primates. Genome Res. 22(4):602–10
98. Peter BM, Huerta-Sanchez E, Nielsen R. 2012. Distinguishing between selective sweeps from standing
variation and from a de novo mutation. PLoS Genet. 8(10):e1003011
99. Pickrell JK, Coop G, Novembre J, Kudaravalli S, Li JZ, et al. 2009. Signals of recent positive selection
in a worldwide sample of human populations. Genome Res. 19(5):826–37
100. Pollard KS, Salama SR, King B, Kern AD, Dreszer T, et al. 2006. Forces shaping the fastest evolving
regions in the human genome. PLoS Genet. 2(10):e168
101. Pollard KS, Salama SR, Lambert N, Lambot M-A, Coppens S, et al. 2006. An RNA gene expressed
during cortical development evolved rapidly in humans. Nature 443(7108):167–72
102. Prabhakar S, Noonan JP, P
¨
a
¨
abo S, Rubin EM. 2006. Accelerated evolution of conserved noncoding
sequences in humans. Science 314(5800):786
103. Pritchard JK, Di Rienzo A. 2010. Adaptation: not by sweeps alone. Nat. Rev. Genet. 11(10):665–67
104. Pritchard JK, Pickrell JK, Coop G. 2010. The genetics of human adaptation: hard sweeps, soft sweeps,
and polygenic adaptation. Curr. Biol. 20(4):R208–15
105. Prud’homme B, Gompel N, Rokas A, Kassner VA, Williams TM, et al. 2006. Repeated morphological
evolution through cis-regulatory changes in a pleiotropic gene. Nature 440(7087):1050–53
106. Przeworski M, Coop G, Wall JD. 2005. The signature of positive selection on standing genetic variation.
Evolution 59(11):2312–23
107. Ram
´
ırez-Soriano A, Nielsen R. 2009. Correcting estimators of θ and Tajima’s D for ascertainment biases
caused by the single-nucleotide polymorphism discovery process. Genetics 181(2):701–10
108. Richards EJ. 2011. Natural epigenetic variation in plant species: a view from the field. Curr. Opin. Plant
Biol. 14(2):204–9
109. Riebler A, Held L, Stephan W. 2008. Bayesian variable selection for detecting adaptive genomic differ-
ences among populations. Genetics 178(3):1817–29
110. Romero IG, Ruvinsky I, Gilad Y. 2012. Comparative studies of gene expression and the evolution of
gene regulation. Nat. Rev. Genet. 13(7):505–16
111. Sabeti PC, Reich DE, Higgins JM, Levine HZP, Richter DJ, et al. 2002. Detecting recent positive
selection in the human genome from haplotype structure. Nature 419(6909):832–37
112. Sabeti PC, Schaffner SF, Fry B, Lohmueller J, Varilly P, et al. 2006. Positive natural selection in the
human lineage. Science 312(5780):1614–20
113. Sabeti PC, Varilly P, Fry B, Lohmueller J, Hostetter E, et al. 2007. Genome-wide detection and char-
acterization of positive selection in human populations. Nature 449(7164):913–18
114. Scheinfeldt LB, Soi S, Thompson S, Ranciaro A, Woldemeskel D, et al. 2012. Genetic adaptation to
high altitude in the Ethiopian highlands. Genome Biol. 13(1):R1
115. Sebat J, Lakshmi B, Troge J, Alexander J, Young J, et al. 2004. Large-scale copy number polymorphism
in the human genome. Science 305(5683):525–28
www.annualreviews.org
•
Detecting Natural Selection in Genomic Data 119
Annu. Rev. Genet. 2013.47:97-120. Downloaded from www.annualreviews.org
by Harvard University on 11/26/13. For personal use only.
GE47CH05-Sabeti ARI 29 October 2013 12:12
116. Shapiro BJ, Alm EJ. 2008. Comparing patterns of natural selection across species using selective signa-
tures. PLoS Genet. 4(2):e23
117. Shriver MD, Kennedy GC, Parra EJ, Lawson HA, Sonpar V, et al. 2004. The genomic distribution of
population substructure in four populations using 8,525 autosomal SNPs. Hum. Genomics 1(4):274–86
118. Simonson TS, Yang Y, Huff CD, Yun H, Qin G, et al. 2010. Genetic evidence for high-altitude adaptation
in Tibet. Science 329(5987):72–75
119. Smith JM, Haigh J. 1974. The hitch-hiking effect of a favourable gene. Genet. Res. 23(1):23–35
120. Smith NGC, Eyre-Walker A. 2002. Adaptive protein evolution in Drosophila. Nature 415(6875):1022–24
121. Stefansson H, Helgason A, Thorleifsson G, Steinthorsdottir V, Masson G, et al. 2005. A common
inversion under selection in Europeans. Nat. Genet. 37(2):129–37
122. Tajima F. 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism.
Genetics 123(3):585–95
123. Tajima F. 1993. Simple methods for testing the molecular evolutionary clock hypothesis. Genetics
135(2):599–607
124. Teshima KM, Coop G, Przeworski M. 2006. How reliable are empirical genomic scans for selective
sweeps? Genome Res. 16(6):702–12
125. Tishkoff SA, Reed FA, Ranciaro A, Voight BF, Babbitt CC, et al. 2007. Convergent adaptation of human
lactase persistence in Africa and Europe. Nat. Genet. 39(1):31–40
126. Tsetskhladze ZR, Canfield VA, Ang KC, Wentzel SM, Reid KP, et al. 2012. Functional assessment of
human coding mutations affecting skin pigmentation using zebrafish. PLoS ONE 7(10):e47398
127. Turner BM. 2009. Epigenetic responses to environmental change and their evolutionary implications.
Philos. Trans. R. Soc. Lond. B 364(1534):3403–18
128. Vanhamme L, Paturiaux-Hanocq F, Poelvoorde P, Nolan DP, Lins L, et al. 2003. Apolipoprotein L-I
is the trypanosome lytic factor of human serum. Nature 422(6927):83–87
129. Vitalis R, Dawson K, Boursot P. 2001. Interpretation of variation across marker loci as evidence of
selection. Genetics 158(4):1811–23
130. Vitti JJ, Cho MK, Tishkoff SA, Sabeti PC. 2012. Human evolutionary genomics: ethical and interpretive
issues. Trends Genet. 28(3):137–45
131. Voight BF, Kudaravalli S, Wen X, Pritchard JK. 2006. A map of recent positive selection in the human
genome. PLoS Biol. 4(3):e72
132. Wang ET, Kodama G, Baldi P, Moyzis RK. 2006. Global landscape of recent inferred Darwinian
selection for Homo sapiens. Proc. Natl. Acad. Sci. USA 103(1):135–40
133. Watterson GA. 1978. The homozygosity test of neutrality. Genetics 88(2):405–17
134. Wiener P, Pong-Wong R. 2011. A regression-based approach to selection mapping. J. Hered. 102(3):294–
305
135. Wright SI, Charlesworth B. 2004. The HKA test revisited. Genetics 168(2):1071–76
136. Yang, Bielawski. 2000. Statistical methods for detecting molecular adaptation. Trends Ecol. Evol.
15(12):496–503
137. Yokoyama S. 1983. Selection for the α-thalassemia genes. Genetics 103(1):143–48
138. Zeng K, Fu Y-X, Shi S, Wu C-I. 2006. Statistical tests for detecting positive selection by utilizing
high-frequency variants. Genetics 174(3):1431–39
139. Zeng K, Shi S, Wu C-I. 2007. Compound tests for the detection of hitchhiking under positive selection.
Mol. Biol. Evol. 24(8):1898–908
140. Zhai W, Nielsen R, Slatkin M. 2009. An investigation of the statistical power of neutrality tests based
on comparative and population genetic data. Mol. Biol. Evol. 26(2):273–83
141. Zhang C, Bailey DK, Awad T, Liu G, Xing G, et al. 2006. A whole genome long-range haplo-
type (WGLRH) test for detecting imprints of positive selection in human populations. Bioinformatics
22(17):2122–28
142. Zhuang Z, Gusev A, Cho J, Pe’er I. 2012. Detecting identity by descent and homozygosity mapping in
whole-exome sequencing data. PLoS ONE 7(10):e47618
120 Vitti
·
Grossman
·
Sabeti
Annu. Rev. Genet. 2013.47:97-120. Downloaded from www.annualreviews.org
by Harvard University on 11/26/13. For personal use only.

GE47-FrontMatter ARI 2 November 2013 9:9
Annual Review of
Genetics
Volume 47, 2013
Contents
Causes of Genome Instability
Andr´es Aguilera and Tatiana Garc´ıa-Muse ppppppppppppppppppppppppppppppppppppppppppppppppp1
Radiation Effects on Human Heredity
Nori Nakamura, Akihiko Suyama, Asao Noda, and Yoshiaki Kodama ppppppppppppppppppp33
Dissecting Social Cell Biology and Tumors Using Drosophila Genetics
Jos´e Carlos Pastor-Pareja and Tian Xu ppppppppppppppppppppppppppppppppppppppppppppppppppppp51
Estimation and Partition of Heritability in Human Populations Using
Whole-Genome Analysis Methods
Anna A.E. Vinkhuyzen, Naomi R. Wray, Jian Yang, Michael E. Goddard,
and Peter M. Visscher pppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppp75
Detecting Natural Selection in Genomic Data
Joseph J. Vitti, Sharon R. Grossman, and Pardis C. Sabeti pppppppppppppppppppppppppppppppp97
Adaptive Translation as a Mechanism of Stress Response
and Adaptation
Tao Pan ppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppp121
Organizing Principles of Mammalian Nonsense-Mediated
mRNA Decay
Maximilian Wei-Lin Popp and Lynne E. Maquat ppppppppppppppppppppppppppppppppppppppp139
Control of Nuclear Activities by Substrate-Selective
and Protein-Group SUMOylation
Stefan Jentsch and Ivan Psakhye ppppppppppppppppppppppppppppppppppppppppppppppppppppppppppp167
Genomic Imprinting: Insights From Plants
Mary Gehring pppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppp187
Regulation of Bacterial Metabolism by Small RNAs
Using Diverse Mechanisms
Maksym Bobrovskyy and Carin K. Vanderpool ppppppppppppppppppppppppppppppppppppppppppp209
Bacteria and the Aging and Longevity of Caenorhabditis elegans
Dennis H. Kim pppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppp233
The Genotypic View of Social Interactions in Microbial Communities
Sara Mitri and Kevin Richard Foster ppppppppppppppppppppppppppppppppppppppppppppppppppppp247
SIR Proteins and the Assembly of Silent Chromatin in Budding Yeast
Stephanie Kueng, Mariano Oppikofer, and Susan M. Gasser pppppppppppppppppppppppppppp275
v
Annu. Rev. Genet. 2013.47:97-120. Downloaded from www.annualreviews.org
by Harvard University on 11/26/13. For personal use only.
GE47-FrontMatter ARI 2 November 2013 9:9
New Gene Evolution: Little Did We Know
Manyuan Long, Nicholas W. VanKuren, Sidi Chen, Maria D. Vibranovski ppppppppppp307
RNA Editing in Plants and Its Evolution
Mizuki Takenaka, Anja Zehrmann, Daniil Verbitskiy, Barbara H¨artel,
and Axel Brennicke pppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppp335
Expanding Horizons: Ciliary Proteins Reach Beyond Cilia
Shiaulou Yuan and Zhaoxia Sun ppppppppppppppppppppppppppppppppppppppppppppppppppppppppppp353
The Digestive Tract of Drosophila melanogaster
Bruno Lemaitre and Irene Miguel-Aliaga ppppppppppppppppppppppppppppppppppppppppppppppppp377
RNase III: Genetics and Function; Structure and Mechanism
Donald L. Court, Jianhua Gan, Yu-He Liang, Gary X. Shaw, Joseph E. Tropea,
Nina Costantino, David S. Waugh, and Xinhua Ji pppppppppppppppppppppppppppppppppppp405
Modernizing the Nonhomologous End-Joining Repertoire:
Alternative and Classical NHEJ Share the Stage
Ludovic Deriano and David B. Roth ppppppppppppppppppppppppppppppppppppppppppppppppppppppp433
Enterococcal Sex Pheromones: Signaling, Social Behavior,
and Evolution
Gary M. Dunny pppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppp457
Control of Transcriptional Elongation
Hojoong Kwak and John T. Lis ppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppp483
The Genomic and Cellular Foundations of Animal Origins
Daniel J. Richter and Nicole King ppppppppppppppppppppppppppppppppppppppppppppppppppppppppp509
Genetic Techniques for the Archaea
Joel A. Farkas, Jonathan W. Picking, and Thomas J. Santangelo ppppppppppppppppppppppp539
Initation of Meiotic Recombination: How and Where? Conservation
and Specificities Among Eukaryotes
Bernard de Massy pppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppp563
Biology and Genetics of Prions Causing Neurodegeneration
Stanley B. Prusiner ppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppp601
Bacterial Mg
2+
Homeostasis, Transport, and Virulence
Eduardo A. Groisman, Kerry Hollands, Michelle A. Kriner, Eun-Jin Lee,
Sun-Yang Park, and Mauricio H. Pontes ppppppppppppppppppppppppppppppppppppppppppppppp625
Errata
An online log of corrections to Annual Review of Genetics articles may be found at
http://genet.annualreviews.org/errata.shtml
vi Contents
Annu. Rev. Genet. 2013.47:97-120. Downloaded from www.annualreviews.org
by Harvard University on 11/26/13. For personal use only.
